
CRISPR-Cas介导的基因编辑工具
2014-07-12 05:42左其生李东张亚妮李碧春
生物技术通报 2014年7期
左其生 李东 张亚妮 李碧春
(扬州大学动物科学技术学院 江苏省动物遗传繁育与分子设计重点实验室,扬州 225009)
随着分子高通量测序、基因芯片和RNA-seq技术的产生、发展,复杂基因组中未知基因功能的探索对生物学的定点研究、临床医学及基因治疗都有着不可替代的前景和作用。从人们对基因功能探索开始,研究者就利用DNA损伤修复的天然机制(自然重组)来实现靶基因的定点修饰,然而自然情况下重组效率极低,且实验重复性差,致使后续试验不能顺利进行。经过科研工作者的不断探索,获得了一些对基因组中特定位点的基因进行靶向敲除或敲入的方法,并取得一定的成效。近年来,人工核酸内切酶(Engineered endonuclease,EEN)方法的出现使基因靶向敲除变得简单,同时也提高了实验的重复性。ENN能够通过特异性的核酸序列介导,利用核酸内切酶对特异性的DNA位点进行酶切,产生双链断裂切口(double strand breaks,DSB),当DNA损伤时,细胞为防止DNA降解启动DNA的自我修复机制:同源重组(homologous recombination,HR)和非同源末端重组(Non-homologous end joining,NHEJ),对断裂的双链核酸进行修复。非同源末端重组修复过程极易产生碱基丢失,错配,在修复过程中会产生不等数目的碱基缺失或者插入,进而改变基因的密码子阅读框,使得翻译后的蛋白质的功能出现差异甚至失活。精准的基因靶向修饰可以清楚的了解目的基因的功能,有利于深入了解疾病的发生机制并制定相应的治疗方案。目前应用比较广泛的ENN主要有锌指核酸酶(Zinc-finger nuclease,ZFN)和转录激活因子样效应物核酸酶(Transcription activator-like effector nuclease,TALEN),然而这两种技术的设计比较复杂,实验过程也较繁琐,而作为第3代ENN技术的Cas9/gRNA系统则是这两年兴起的一种快速基因靶向敲除技术,因其具有实验过程简单、耗时短和工作量小等优势,正逐渐地取代ZFN和TALEN技术。目前研究人员已经利用Cas9系统在小鼠、斑马鱼和果蝇等动物中成功的构建了基因敲除模型,并在HEK293和iPS等细胞中实现了基因的敲除并产生了稳定的细胞株,实现了基因的定点修饰及其功能研究。因此,本文就Cas9系统的作用原理、设计步骤及其应用进行系统的综述。
1 CRISPR-Cas的发现、结构及作用原理
规律性重复短回文序列簇(clustered regularly interspaced short palindromic repeats,CRISPRs)是细菌和古细菌在不断进化的过程中获得一种适应性免疫防御机制,通过小片段的RNA介导对入侵的核酸进行靶向定位并通过Cas酶对核酸进行酶切、降解[1]。研究者根据CRISPR-Cas系统的特性对此系统进行改造产生了第3代人工核酸内切酶技术。利用此技术能够对目的基因进行靶向的敲除(Knock out)或敲入(Konck in)。
1.1 CRISPR-Cas的发现
1987年,日本学者在研究细菌中编码碱性磷酸酶基因时发现,在这个基因的编码区附近有一小段DNA片段包含了大量的重复序列[2],直到2002年,科学家才将其正式命名为 Clustered regularly interspaced short palindromic repeats(CRISPR)[3]。研究发现,大约有40%的细菌中含有CRISPR位点,但是不同细菌种属在该位点的基因序列具有特异性[4]。2005年,科研人员发现CRISPR中的重复序列与细菌的外源核酸入侵的免疫过程有关,其功能类似于真核生物的RNAi干扰方式。此后,科研人员陆续的探索了CRISPR的功能和机制,并将其应用于生物、医药、农业等领域,使之成为继ZFN和TALEN之后的又一重要的基因靶向修饰技术。
1.2 CRISPR/Cas9 的结构
CRISPR是一种特殊的DNA序列重复家族,是细菌和古细菌在不断进化的过程中产生的一种特异性免疫防御机制。CRISPR-Cas主要分为I、II、III 3个类型,Type I系统分布于细菌和古细菌中,组分复杂,核心蛋白元件为Cas3 蛋白,该蛋白具有DNA核酸酶和解旋酶功能,在防御外源核酸入侵时,多个Cas蛋白与crRNA形成复合物CASCAD(CRISPR associated complex for antivirus defense),在crRNA的介导下与外源核酸特异性结合,Cas3启动核酸酶作用,对外源核酶进行酶切,降解。Type II系统主要分布于细菌中,尤其是产链脓杆菌。该系统组分简单,核心蛋白元件为Cas9蛋白,Cas9蛋白与成熟的crRNA结合形成复合物即可对外源的核酸进行酶切,降解。Type III系统主要分布于古细菌中,在细菌中比较少见。该系统的核心蛋白元件为Cas10,其作用于Type I的CASCAD类似,主要参与crRNA的成熟以及外源核酸的降解。根据系统的靶标对象的不同可将Type III分为Type III A和Type III B,前者的靶标对象为mRNA,后者为DNA[5]。
由于II型CRISPR的系统组分相对简单,因此目前研究多集中于II型CRISPR系统。II型CRISPR/Cas的组成,如图1所示,首先是5'端的tracrRNA,此tracrRNA能够与3'端的spacers序列转录剪切成熟后的crRNA通过碱基互补配对形成双链RNA,tracrRNA/crRNA二元复合体指导Cas9蛋白在crRNA引导序列靶标的特定位点剪切双链DNA;其次是Cas蛋白的编码序列 Cas9、Cas1、Cas2和Csn2,不同的亚型蛋白分别在DNA的复制、转录和翻译过程中发挥不同的作用,其中Cas9具有核酸酶的作用。Cas9包含HNH核酸酶结构域和RuvC-like 结构域,HNH核酸酶结构域能够剪切与crRNA互补的序列而RuvC-like 结构域则剪切非互补的序列;3'端为CRISPR 基因座,由启动子区域和众多的重复序列(21-48 bp左右,序列并不严格保守)和间隔序列组成,重复序列中的回文序列在Cas蛋白形成核糖核蛋白复合物的过程中起着重要的作用。间隔序列决定了CRISPR系统的特异性,间隔序列的差异来源于外源入侵的噬菌体和质粒,一般在21-72 bp,不同的间隔序列能够记录不同的噬菌体或者质粒的入侵的时间和顺序。

图1 II型CRISPR/Cas结构示意图
1.3 CRISPR-Cas 系统的作用原理
任何一种生命体都可以通过某一RNA介导的防御机制来保护自身的基因组免受外源核酸的干扰和破坏,如真核生物中miRNA干扰现象,CRISPR-Cas系统则是细菌和古细菌在不断进化的过程中产生的特异的免疫防御机制,当外源核酸入侵时对其靶向定位并进行降解,其作用过程主要分为3步:首先CRISPR间隔序列的获得和整合;其次,CRISPR基因座的表达;再次,CRISPR-Cas功能的发挥,即对外源核酸的干扰[6]。
当噬菌体或者外源质粒初次入侵时,其核酸的一小段DNA片段将被动的整合到宿主的基因组中,整合部位位于 CRRSPR的5'端的初始的重复序列中形成间隔序列,而噬菌体或质粒中与之对应的序列则为原间隔序列(Protospacer),而原间隔序列的5'端和3'端延伸的几个序列是非常保守的,称之为原间隔序列临近基序(Protospacer adjacent motifs,PAM),PAM的一般形式为NGG,其作用是将间隔序列定位于入侵的噬菌体或质粒的DNA序列中,即宿主对入侵的外源核酸进行扫描,在其DNA序列中定位若干个PAM,并将PAM 5'端或3'的序列定义为新的原间隔序列并被剪切后陆续的整合到CRRSPR系统5'端新合成的两个重复序列之间(图2)。间隔序列的产生与Cas2的功能是分不开的,大量研究发现所有的CRISPR系统中都含有 Cas2基因编码区,表明Cas2在CRISPR系统作用的过程中起着非常重要的作用,研究结果也表明Cas2参与了新的原间隔序列的形成。
正常情况下细菌中的CRISPR的表达水平较低且相对恒定。当噬菌体或外源质粒再次入侵时,CRISPR会被迅速的诱导上调表达。重复序列和间隔序列在前导序列的启动下进行转录形成pre-crRNA,pre-crRNA通过剪切形成成熟的cr-RNA。cr-RNA由两部分组成即间隔序列和侧翼的部分重复序列。cr-RNA与tracrRNA形成双链的RNA,对入侵的外源核酸进行靶向的定位并介导Cas9核酸酶对外源核酸进行切割降解。目前发现并非只有cr-RNA与外源DNA完全匹配的才会被酶切,随着该技术的不断发展,研究表明Cas9技术在实验过程中存在脱靶效应,即cr-RNA与外源DNA不完全匹配的情况下序列也有可能被酶切。
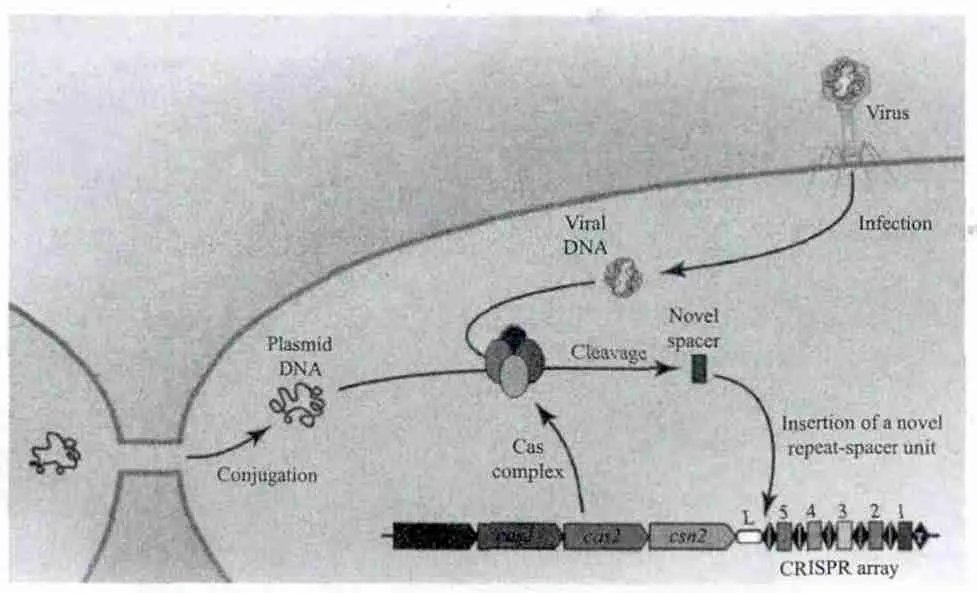
图2 II型CRISPR-Cas系统原间隔序列形成过程
2 Cas9/gRNA的体外构建步骤
研究人员发现Cr-RNA与tracrRNA形成的双链RNA(guide RNA,gRNA)介导的双链DNA剪切系统的核心,人为的改造这一双链RNA可以了解细菌Cas9对不同物种的DNA系列进行任意的切割,gRNA介导Cas9对靶基因进行切割并形成DSB是实现基因修饰的基础。Hwang等[7]利用人工合成的sgRNA指导了细菌蛋白Cas9对斑马鱼胚胎基因进行了修饰,其效率类似于ZFN和TALEN技术。Cas9/gRNA系统体外构建的一般步骤(图3):(1)从已知的序列中通过PAM进行定位寻找合适的靶位点并合成gRNA,在设计靶位点应尽量靠近第一外显子附近,这样能够获得较高活性的gRNA;(2)将合成好的gRNA与Cas9序列构建重组质粒。目前在构建重组质粒过程中有两种主要方案:一是将gRNA与Cas9连入同一个质粒载体中并以双启动子启动,载体中携带荧光报告基因(用于流式细胞仪筛选)或抗性基因(用于药物筛选)。二是将gRNA与Cas9分别连载不同的载体上,两个载体携带有不同的荧光报告基因或抗性基因;(3)对构建好的重组质粒进行活性检测,即敲除活性的计算;(4)挑选活性较高的敲除质粒或者质粒对通过转染或者显微注射的方法导入培养的细胞或者生物体内进行基因组定点编辑操作;(5)利用测序、RFLP等方法检测基因组定点修饰的结果。与ZFN、TALEN不同的是,Cas9系统的gRNA的设计比较简单,而第3步的活性检测则关系到后期基因的敲除效率,为Cas9系统实验过程中的关键一步。

图3 Cas9/gRNA系统体外构建步骤流程图
2.1 gRNA的设计
sgRNA是体外人工合成的一小段单链核糖核苷酸,约19-23 bp,其中PAM和5'端的8-12 bp的seed seqence 对靶向切割双链DNA最为重要。PAM的存在形式主要为NGG,其中N为5'端,PAM的主要作用是对目标DNA序列中的原间隔序列进行定位,使sgRNA能够与靶位点进行特异性结合并介导Cas9蛋白识别并在其5'端实现切割。除了PAM和seed seqence之外的5'端的其他碱基序列在对靶位点的识别时容易脱靶,这也是Cas9在试验过程中出现脱靶效应的原因;在试验过程中为方便后期突变位点的检测,选择PAM 5'端存在酶切位点的原间隔序列作为gRNA,可以通过酶切鉴定基因修饰的结果;研究还发现在设计sgRNA时,如果DSB的侧翼存在重复序列,在进行非同源末端重组时能够精确的介导断裂位点碱基的缺失。
综上,sgRNA的设计原则有以下几点:(1)选择PAM(NGG)5'端的一段碱基序列作为原间隔序列,即敲除的靶位点。(2)选择的序列必须在全基因组中进行比对,原间隔序列必须唯一,否则会对其他基因进行敲除而出现错误的实验结果。(3)优先选择DSB位点的侧翼存在重复序列;(4)PAM的5'端尽量存在酶切位点。
2.2 gRNA的活性检测
对设计好的sgRNA进行活性检测以获得突变效率较高的sgRNA用于后期的实验。目前主要采用的方法有限制性内切酶法、非配对内切酶法和SSA活性检测。
2.2.1 限制性内切酶法 生物体内存在着一些内切酶类,它们能够特异性的识别DNA双链中的特定序列并对其进行切割,这些酶能够降解外源的DNA使之失活却对本身固有的DNA没有影响,所以称之为限制性内切酶。根据限制性内切酶的这种特性,设计sgRNA时应选择在PAM的5'端存在酶切位点的原间隔序列(特异性的靶位点)。在Cas9/sgRNA靶点位置中间序列中有限制性内切酶切位点,如HindIII,如果通过Cas9/sgRNA发生突变,这个位点将可能被破坏,而不能被HindIII酶切;同时野生型的细胞则可被HindIII酶切。可采用电泳的方法估计突变效率,以突变效率的高低来衡量sgRNA的活性。Wang[8]等在设计针对Tet1、Tet2和Tet3这3个基因的sgRNA时分别引入ScaI、EcoR V和XhoI酶切位点,利用相应的内切酶对转染了Cas9/sgRNA细胞的DNA进行酶切后,其突变效率分别为36%、48%和36%。
2.2.2 非配对内切酶法 T7 核酸内切酶 I(T7 endonuclease I,T7E1) 能够识别不完全配对DNA并对其进行切割,还能对十字型结构 DNA、Holliday结构或交叉 DNA、异源双链DNA 进行识别和切割,该酶切割错配碱基 5'端的第1、第2或第3个磷酸二酯键。如果通过Cas9/sgRNA发生突变,将基因组DNA做PCR,将相对应的PCR产物与野生型DNA的PCR产物等量混合,并退火杂交,将产生非配对DNA片段,将能被非配对内切酶T7E1剪切。若没有发生突变,将产生配对DNA片段,而无法被非配对内切酶T7E1剪切。用电泳的方法估计突变效率,以突变效率的高低来衡量sgRNA的活性。Chang等[9]利用此法估算Cas9/sgRNA和TALEN技术处理的突变效率,结果表明Cas9/ sgRNA的突变效率比TALEN技术的突变效率要高10%-15%。
2.2.3 SSA活性检测 SSA活性检测原理如图4所示:一个终止子插入luciferase(GFP)的编码区中央(红色标记),luciferase(GFP)就会失去活性。为检测Cas9/sgRNA剪切活性,将一个Cas9/sgRNA的靶点位置序列插在终止子后(蓝色标记)。在Cas9/ sgRNA的作用下,靶点位置产生DSB,细胞通过同源重组方式修复DNA,形成一个有活性的luciferase。通过与对照组的比值变化就可反应Cas9/sgRNA剪切的活性水平。Chang[9]在对斑马鱼胚胎基因组进行修饰时以GFP为报告基因进行了SSA活性检测,结果显示Cas9/sgRNA处理组的荧光强度是对照组的9倍。
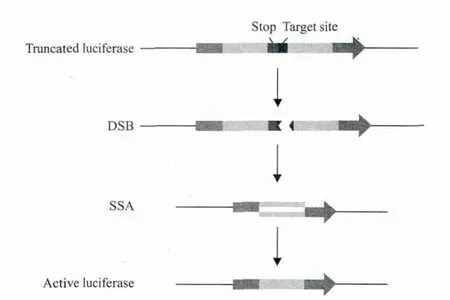
图4 SSA活性检测原理示意图(颜色标识见电子版)
3 Cas9/sgRNA系统的应用
虽然CRISPR/Cas的发现可以追溯到1987年,但是这种技术的应用和发展却是近两年才开始的,目前这项技术已经被广泛应用于动物的细胞水平或者个体水平的基础研究,也已经在HEK293细胞、iPS等细胞中产生了稳定的敲除细胞株。另外,科研人员利用显微注射等方法获得小鼠、大鼠和斑马鱼等模式动物的敲除个体并顺利产生后代。
3.1 Cas9/sgRNA系统细胞水平进行基因的修饰
目前,科研人员已经利用产链脓杆菌的typeII型系统改造的Cas9系统在人类细胞,小鼠及斑马鱼等物种中完成基因修饰并取得良好结果。
Jinek[10]课题组以人类细胞为实验材料进行基因的靶向修饰,试验中筛选的细胞顺利产生DSB,说明Cas9系统可用于人类细胞。他们对Cas9系统进行了改造,改造过程中发现TracrRNA是Cas9系统具有活性必不可少的组分,作者还详细阐述了Cas9系统的切割原理以及切割的具体位置;试验还表明gRNA的表达对Cas9的活性是必不可少的,而且gRNA的3'端的适当延伸能够显著增加Cas9系统的活性。Jinek等的试验结果为后续的Cas9系统的研究和应用奠定了基础,为基因靶向修饰提供了一个除ZFN和TALEN之外的另一个宝贵的实验工具,这为基础医学尤其是基因治疗提供了新思路、新方法、新途径。
随后研究人员对Cas9系统中不同组分对对实验的影响进行了探索。Cho[11]团队以人的细胞为试验材料,通过试验证明在共转染体系中gRNA的浓度的增加能够提高基因的敲除效率,最高效率达到33%。Mail等[12]改造了Cas9系统以用于人类基因组编辑,并顺利地在HEK 293和K652细胞中实现基因的靶向敲除。Cong等[13]利用Cas9系统在小鼠的nero2A细胞中的Th基因以及HEK293细胞的EMX1基因,PVALB基因实现定点敲除。Cong和mali通过试验表明gRNA在结构上越接近crRNA和tracrRNA复合体,则在试验过程中能够获得较高的敲除效率。
目前利用Cas9进行基因敲除的技术已经日渐成熟,利用此技术在细胞中可实现单基因或者多基因的敲除。Mali等[12]利用此技术在人的细胞中实现了基因的敲除,靶向敲除效率分别为HEK 293为10%-25%,K562为8%-13%,iPS为2%-49%;Wang等[8]通过共转染的方法在小鼠的胚胎干细胞中对Tet1、Tet2、Tet3、Uty和Sry等基因中实现了单基因、双基因及多基因的敲除,敲除效率达40%左右,并通过测序、RFLP及DNA印迹等方法进行了验证。Cong等[13]也在人和小鼠的细胞中也实现了多基因的同时敲除。利用多基因敲除试验的成功将有利于科研人员在体内研究冗余基因以及上位基因的功能。
3.2 Cas9/sgRNA系统个体水平进行基因的修饰
自20世纪80年代第一只基因敲除小鼠的产生,科研工作者一直致力于基因敲除模型的制备,期间虽然ZFN和TALEN也曾一时兴起,然而这两种方法耗时耗力,过程也很繁琐。Cas9系统的产生为基因敲除模型的制备提供了新方法。
Friedland等[14]以线虫为生物模型,通过显微注射的方法向线虫胚胎细胞中注射Cas9/gRNA质粒,实现基因在个体水平上的基因敲除,敲除效率约为88%,为保证这种方法的顺利使用,作者还介绍了一种新的检测基因突变的方法——高分辨率融化分析(High-resolution melt analysis)。Hwang 等[7]利用Cas9/ gRNA通过对斑马鱼胚胎 drd3、gsk3b基因进行修饰,获得了这两个基因位点的突变的突变体;Li等[15]利用Cas9系统在小鼠的个体上实现了基因的靶向修饰,通过显微注射的方法将Cas9以及gRNA的mRNA共注射进小鼠的胚胎干细胞对Uhrf2基因进行定点修饰,获得了Mc3R、Mc4cRL两只双基因敲除小鼠。Wang等[8]通过显微共注射mRNA的方法获得了Tet1、Tet2的单基因敲除小鼠以及双基因敲除小鼠,并在其后代中仍然为敲除阳性。利用Cas9技术产生基因敲除模型能够很好的应用于基因治疗以及家畜育种过程中。
4 存在的问题
根据实验的需要以及gRNA的设计原则设计出符合实验需求且特异性较高的gRNA,并能在活性检测过程中具有较高的活性(即较高的敲除效率)为试验难点之一。
避免试验过程中Cas9系统的脱靶效应。虽然Wang[8]对Cas9系统的脱靶效应进行了研究,结果表明无脱靶效应,但是由于物种之间存在差异以及所设计的gRNA之间有差别,因此Cas9系统的脱靶效应有待于进一步研究。
Cas9系统在高度分化的细胞、细胞系以及个体上都有较为理想的试验结果,但是在未建系的干细胞上尚未有成功的先例,尤其是在转染Cas9/gRNA质粒或者mRNA后如何筛选出阳性克隆进行后续试验这一问题有待解决。
Cas9系统的毒性问题。Cas9为细菌蛋白,目前尚不清楚其在其他物种的细胞中作用是否会产生毒性。Wang等在他们的试验中对Cas9的用量进行了梯度实验,结果表明,在适当的范围内提高Cas9的浓度有利于提高基因的敲除效率,但是200 ng的浓度不能证明对其他物种也适用。因此Cas9系统的毒性问题有待于进一步解决。
5 展望
II型CRISP/Cas应用为一种新型的基因靶向修饰技术,虽然这种应用尚处于起步阶段,但是相比于ZFN、TALEN技术的耗时、耗力以及设计繁琐等缺点而言Cas9以其设计简单,耗时短,实验操作性强等优势而被科研人员广泛使用。一般而言,以小鼠为试验材料,Cas9系统最快可在6-8周内产生基因敲除模型,这就大大的节省了试验时间而且成功率高。ZFN和TALEN以FoxI酶为核心,而Cas9系统则以Cas9酶为核心,在修饰过程中Cas9系统在gRNA的介导下的特异性比ZFN、TALEN更强,能够更加准确的对基因进行修饰;当需要针对基因组中不同的切割位点时,ZFN、TALEN需要重新设计,而Cas9/gRNA系统只要重新合成gRNA即可。这些是ZFN、TALEN技术所达不到的。
虽然目前II型CRISP/Cas的研究尚属崭新领域,但是此技术将成为基因工程研究的一种新工具,虽然关于CRISP/Cas的系统具体机制不甚了解,部分问题有待于进一步解决,但是我们相信随着科研人员对CRISP/Cas系统研究的不断深入,CRISP/Cas系统将更好的帮助科研人员了解基因的功能,探索基因组的奥秘。
[1] Lillestøl R, Redder P, Garrett RA, et al. A putative viral defence mechanism in archaeal cells [J] . Archaea, 2006, 2(1):59-72.
[2] Mahfouz MM, Li LX, Shamimuzzaman M, et al. De novo-engineered transcription activator-like effector(TALE)hybrid nuclease with novel DNA binding specificity creates double-strand breaks [J] .Proc Natl Acad Sci USA, 2011, 108(6):2623-2628.
[3] Coffey A, Ross RP. Bacteriophage-resistance systems in dairy starter strains, molecular analysis to application [J] . Antonie van Leeuwenhoek, 2002, 82(1-4):303-321.
[4] Wei CX, Liu JY, Yu ZS, et al. TALEN or Cas9 - rapid, efficient and specific choices for genome modifications [J] . Genet Genomics,2013, 40(6):281-289.
[5] Makarova KS, Haft DH, Barrangou R, et al. Evolution and classification of the CRISPR- Cas systems [J] . Nat Rev Microbiol, 2011,9(6):467-477.
[6] Marraffini LA, Sontheimer EJ. CRISPR interference:RNA-directed adaptive immunity in bacteria and archaea [J] . Nat Rev Genet,2010, 11(3):181-190.
[7] Hwang WY, Fu YF, Reyon D, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system [J] . Nature Biotechnology,2013, 31(3):227-229.
[8] Wang HY, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering [J] . Cell, 2013, 153(4):910-918.
[9] Chang NN, Sun CH, Gao L, et al. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos [J] . Cell Res, 2013, 23(4):465-472.
[10] Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J] .Science, 2012, 337(6096):816-821.
[11] Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease [J] . Nature Biotechnology, 2013, 31(3):230-232.
[12] Mali P, Yang LH, Esvelt KM, et al. Church GM. RNA-guided human genome engineering via Cas9 [J] . Science, 2013, 339(6121):823-826.
[13] Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems Science [J] . Science, 2013, 339(6121):819-823.
[14] Friedland AE, Tzur YB, Esvel KM, et al. Heritable genome editing inC. elegansvia a CRISPR-Cas9 system [J] . Nature Methods,2013, 10(8):741-743.
猜你喜欢
舰船科学技术(2022年11期)2022-07-15
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
数学小灵通(1-2年级)(2020年11期)2020-12-28
中国(俄文)(2020年8期)2020-11-23
西藏农业科技(2019年3期)2019-11-04
小学生学习指导(低年级)(2019年3期)2019-04-22
现代园艺(2018年3期)2018-02-10
上海农业学报(2017年3期)2017-04-10
