
SPE-HPLC 法检测食品中多种痕量雌激素
2014-07-08 02:16黄璐张勇民郑琦
江汉大学学报(自然科学版) 2014年1期
黄璐,张勇民,郑琦
(江汉大学交叉学科研究院,湖北武汉430056)
SPE-HPLC 法检测食品中多种痕量雌激素
黄璐,张勇民,郑琦*
(江汉大学交叉学科研究院,湖北武汉430056)
建立了固相萃取-高效液相色谱(SPE-HPLC)法同时测定食品中炔雌醇、己烯雌酚、壬基酚、苯甲酸雌二醇残留量的方法。样品以乙腈超声萃取、离心,提取液经C18固相萃取柱浓缩净化后,采用高效液相色谱仪进行分析。对固相萃取条件进行了优化,4种组分均得到了良好的分离,方法的检出限为6~12μg/kg,加标回收率为80.3%~107.8%,工作曲线的相关系数为0.998 8~0.999 8,相对标准偏差是0.124%~1.72%。结果表明,方法灵敏度高,分离效果好,样品处理简单、可减少杂质干扰,能够满足食品中雌激素分析的需要。
固相萃取;高效液相色谱法;痕量雌激素;食品检测
近年来,雌激素残留污染问题已引起全世界广泛关注,尤其是人工合成的雌激素具有强且持久的蛋白质同化作用,致使动物超常态生长,大幅度提高动物养殖经济效益[1]。当前,养殖业中雌激素的过量使用,导致食品中雌激素残留,进入人体后扰乱体内激素平衡,影响生殖系统、神经系统,导致发育异常、性早熟等,甚至具有潜在致癌、致畸风险[2]。
目前,对食品中的雌激素残留分析检测的方法主要有色谱法[3]、色谱-质谱联用[4]、光谱法[5]、免疫分析法等[6-7]。雌激素在实际样品中的含量很低且基质复杂,液液萃取等传统的提取方法虽然成本低但操作复杂、富集倍数不够、有机溶剂用量大,并且在提取过程中有机溶剂对样品中杂质成分的无区别提取常对目标物质的检测造成干扰。近年来发展起来的固相萃取(Solid phase ex⁃traction,SPE)采用固体物质作为萃取剂,利用其选择性吸附与洗脱的原理对样品进行富集、分离和纯化,操作简单,富集倍数高,对环境污染小,能够得到较为纯净的待测物,减少了样品中复杂基质的干扰。笔者建立了SPE-HPLC联用的方法,用SPE法对样品进行预处理,除去部分杂质减少干扰,并经过一定的富集浓缩后以高效液相色谱(HPLC)再次检测,提高了灵敏度。
1 实验部分
1.1 仪器与试剂
1.1.1仪器DIONEX高效液相色谱仪(配有GP50型梯度泵,TCC-100柱温箱,UVD170U型紫外检测器),美国戴安公司;色谱柱:Thermo C18柱(250 mm×0.46 mm,5μm);旋转蒸发仪RE-52AA,上海亚荣生化仪器厂;KQ-250超声波提取仪,昆山市超声仪器有限公司;微孔滤膜(孔径0.45μm);800型低速离心沉淀机,中国深圳国华仪器;DENVER instrument电子天平;SPE装置(Supelco);C18固相萃取柱(3m L,Supelco)。
1.1.2试剂对照品己烯雌酚(批号F1219049)、苯甲酸雌二醇(批号45131)购于阿拉丁试剂(上海)有限公司,炔雌醇、壬基酚由德国进口;甲醇(AR)、乙腈(AR、LC)、磷酸二氢钠(AR)均购于国药集团化学试剂有限公司;实验用水为双蒸水。精密称取炔雌醇、己烯雌酚、壬基酚、苯甲酸雌二醇标准品各50 mg,用甲醇定容至50 m L,得到1 mg/m L的贮备液,存放于冰箱备用,实验时根据需要稀释至所需浓度。样品为超市购得的奶粉、鲫鱼和鳊鱼。
1.2 色谱条件
流动相:乙腈—0.043mol/L磷酸二氢钠缓冲液(用磷酸调至pH为5)[8](梯度洗脱:1~5 min乙腈—磷酸二氢钠缓冲液55:45,5~15min乙腈—磷酸二氢钠缓冲液85:15,15~20 m in乙腈—磷酸二氢钠缓冲液55:45);流速1mL/min;柱温30℃;检测波长224 nm;进样量20μL。
1.3 样品处理
称取5.0 g样品于50 m L烧杯中,加10 m L乙腈混匀,超声提取10min,4 000 r/min离心10min,取上清液静置,滤渣再加10 m L乙腈超声提取10 min后离心,合并两次上清液,旋转蒸发至近干[9]。蒸干后的残余物用20%乙腈水溶液溶解用于下一步实验。样品溶液以2~3m L/min的速度通过预先活化好C18固相萃取柱(依次用12m L甲醇和4mL蒸馏水流经C18固相萃取柱进行活化),上样后用体积浓度40%的甲醇水溶液5 m L淋洗去除部分杂质,最后用3mL甲醇洗脱,收集洗脱液,0.45μm滤膜过滤,以HPLC法检测。
2 结果与分析
2.1 固相萃取条件的优化和讨论
固相萃取的操作过程如下图1(控制流速为2~3m L/min):
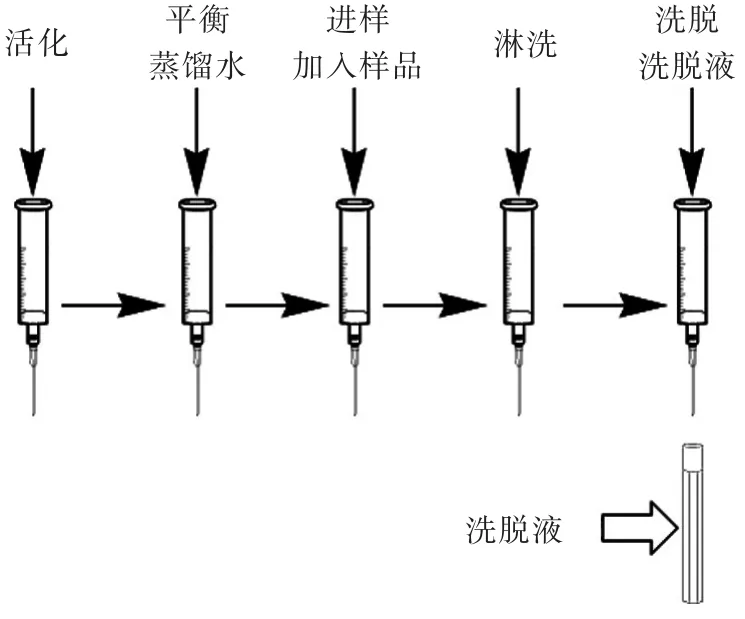
图1 固相萃取操作过程Fig.1 Solid phase extraction process
2.1.1活化液体积的选择C18固相萃取柱一般是用甲醇活化后用蒸馏水平衡,活化是否完全直接关系到进样时萃取柱对目标物的吸附,本实验探索了活化液甲醇的用量对活化效果的影响。依据图1过程操作,改变活化液甲醇体积,收集最终的洗脱液,用HPLC检测。检测结果显示,当活化液甲醇体积增加为12m L时检测值最高,进一步增加至14、16mL时反而检测值下降。由此可知,当活化液甲醇体积较少时,C18固相萃取柱填料未能充分活化,影响目标物在萃取柱上的吸附,当超过一定体积后,萃取柱中大量的甲醇残留有可能改变萃取柱的极性,影响目标样品在萃取柱上的吸附。所以最终选择用12m L甲醇活化C18固相萃取柱。
2.1.2上样时溶液极性的选择将各组分标准混合样分别用20%、40%、60%、80%和100%乙腈水溶液溶解,等体积浓度的溶液上样,按图1过程操作,收集上样后的流出溶剂,用液相色谱检测。检测结果显示40%、60%、80%和100%乙腈水溶液溶解的样品在上样后均能在吸附后的流出溶剂中检出部分目标物,说明样品并未完全吸附。20%乙腈水溶液溶解的样品在上样后的流出溶剂中未检出任何目标物,说明目标物几乎被完全吸附。所以选择上样溶液的溶剂为20%乙腈水溶液。
2.1.3洗脱液的选择选择甲醇、乙腈、乙酸乙酯、正己烷为洗脱液,按图1过程操作,收集洗脱液,用HPLC检测,对比回收率。检测结果显示,甲醇和乙腈作为洗脱液时对样品的回收率影响改变不大,乙酸乙酯和正己烷由于极性较弱,能够被固相萃取柱吸附,不适用于作为C18固相萃取柱的洗脱溶剂。甲醇作为洗脱溶剂洗脱回收率较乙腈高,所以选取甲醇为洗脱溶剂。
确定洗脱溶剂的种类后,其他条件不变,改变洗脱液的体积,按图1过程操作,收集洗脱液,用HPLC检测。检测结果显示,3m L甲醇可以将目标物完全洗脱。
2.1.4淋洗液的选择由于甲醇对于脂肪、蛋白质等物质的溶出性较好,所以选用不同比例的甲醇作为淋洗溶剂。分别选用0%、10%、20%、30%、40%、50%、60%、70%、80%、90%和100%甲醇水溶液为淋洗溶剂,按图1过程操作,收集淋洗液,用HPLC检测。检测结果显示,70%的甲醇水溶液作为淋洗溶液时部分炔雌醇和己烯雌酚被洗脱,80%的甲醇水溶液作为淋洗溶液时部分壬基酚和苯甲酸雌二醇被洗脱,70%以下的甲醇水溶液作为淋洗液时并未洗脱任何目标物。因此我们可以根据食品性质的不同,选取60%及以下浓度的甲醇水溶液作为淋洗液用于杂质组分的淋洗。
2.2 线性范围、检出限、定量限
将各组分不同浓度的标准液用HPLC检测,以3倍信噪比(S/N)计算检出限,以10倍信噪比(S/N)计算定量限,并根据外标法在线性范围内取5个浓度点做出标准曲线。结果见表1。从表1可以看出4种激素类组分的线性关系良好,相关系数为0.998 8~0.999 8,检出限为6~12μg/kg,定量限为25~47μg/kg。

表1 4种激素类药物的线性方程、检出限及定量限Tab.1 The linear equation,detection lim itand thequantitative lim itof the four estrogens
2.3 方法的回收率与精密度
用空白奶粉和鱼肉试样进行添加回收率和精密度实验。精密称取样品5.0 g,分别添加浓度为1μg/m L的4种激素标准溶液1m L,摇匀,按1.3项下样品处理方法操作,每个添加水平测定3次,计算平均加标回收率和相对标准偏差(RSD),结果如表2所示。从表2可以看出,各组分的回收率为80.3%~107.8%。由平行实验测得的数据计算得到相对标准偏差为0.124%~1.72%,可见本实验的准确度与精密度良好。空白和加标样品色谱图见图2、图3。
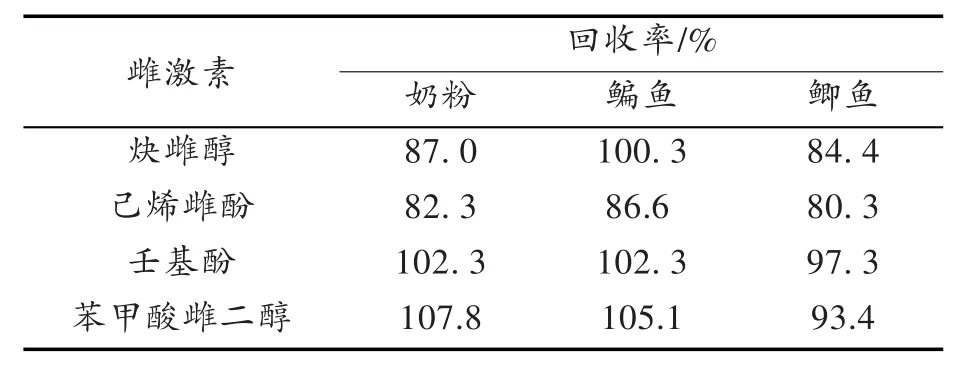
表2 4种激素在不同样品中的加标回收率Tab.2 The recovery of the fou r estrogens in d ifferen t sam p les
2.4 样品分析
采用本方法对超市购得的奶粉、鳊鱼和鲫鱼进行检测,所有样品均低于本方法检出限,未检测出4种待测雌激素成分。各样品在检测过程中均得到良好分离,未发现其他明显的干扰。

图2 样品空白色谱图Fig.2 Ch rom atogram s of b lank sam p le

图3 样品加标色谱图Fig.3 Ch rom atogram s of sam p lew ith standard
3 结论
应用建立的SPE-HPLC法同时测定食品中炔雌醇、己烯雌酚、苯甲酸雌二醇、壬基酚4种激素类药物残留,各组分的加标回收率为80.3%~107.8%,相对RSD为0.124%~1.72%。实验结果表明,该方法可对样品中痕量物质进行净化、富集浓缩,提高了检测的灵敏度,操作简单、快速、成本低,应用于食品中激素类成分残留量的检测,可以得到满意的结果。
[1]牛晋阳,孙焕,李莹莹.高效液相色谱-串联质谱法测定猪肉中10种类固醇激素残留[J].食品科学,2010,31(4):230-232.
[2]许婷婷.动物肌肉中同化激素类药物残留的HPLC分析[D].无锡:江南大学,2009.
[3]迂君,张秀丽,张玉黔.高效液相色谱法测定牛奶中雌激素残留量[J].中国卫生检验杂志,2007,17(6):1139-1140.
[4]耿金培,曹鹏,徐英江,等.超高效液相色谱串联质谱法检测肉制品中八种雌激素残留量[J].中国食品,2011(10):85-87.
[5]罗板鑫,陈昌云.一种简单快速的己烯雌酚定性检测方法研究[J].中国教育技术装备,2012(6):126-127.
[6]郁倩.动物性食品和水中雌激素残留污染检测的研究[D].无锡:江南大学,2008.
[7]王瑶,周建华,张鸿雁,等.食品中己烯雌酚的检测方法[J].食品工业科技,2012,33(3):373-375.
[8]程盛华,杨春亮,查玉兵,等.高效液相色谱法测定鸡肉中己烯雌酚[J].理化检验-化学分册,2008,44(1):15-16.
[9]康莉,仲岳桐,陈晓春,等.固相萃取-高效液相色谱法测定肉中盐酸克伦特罗[J].中国卫生检验杂志,2003,13(1):53-54.
Determ ination of Trace Estrogens in Food by SPE-HPLC
HUANG Lu,ZHANG Yong-min,ZHENGQi
(Institute for Interdisciplinary Research,Jianghan University,Wuhan 430056,Hubei,China)
The residuesof Ethinylestradiol,Diethylstilbestrol,4-Nonylphenoland Estradiolben⁃zoate in food were determined simultaneously by SPE-HPLC.The sampleswere ultrasonic extract⁃ed with acetonitrile,centrifuged,concentrated and purified by C18solid phase extraction column,an⁃alyzed with the HPLC method.This study optimized the conditions of SPE,and four components werewell separated.The detection limitof themethod was ranged from 6 to 12μg/kg.The recovery rateswere 80.3%~107.8%.The calibration curveswere linearwith correlation coefficient ranged from 0.998 8 to 0.999 8.The relative standard deviationswere 0.124%~1.72%.Thismethod has high sensitivity,fine separability,simple sample preparation,and can reduce the inteference of im⁃purities,which is satisfied with the estrogen analysis.
solid phase extraction;HPLC;trace estrogen;food detection
TS207.5;O657.7
A
1673-0143(2014)01-0055-04
(责任编辑:叶冰)
2013-02-01
湖北省科技厅项目(2011CDC130);武汉市科技局项目(201220837304-3)
黄璐(1989—),女,硕士生,研究方向:化学工程。
*通信作者:郑琦(1962—),女,教授,研究方向:食品、药品分析化学。E-mail:zq_1101@sina.com
猜你喜欢
食品工程(2020年4期)2021-01-20
化工管理(2020年26期)2020-10-09
中国油脂(2020年3期)2020-04-10
无机化学学报(2016年8期)2016-12-06
化学分析计量(2016年1期)2016-03-14
水生生物学报(2015年1期)2015-11-05
山西大同大学学报(自然科学版)(2015年1期)2015-01-22
食品科学(2014年21期)2014-03-08
中国造纸(2014年1期)2014-03-01
无机化学学报(2014年8期)2014-02-28
