
钙网蛋白介导的线粒体功能异常参与心肌细胞肥大过程
2014-06-27 10:36:02单虎,魏瑾,张明,林琳,闫蕊,张蓉
西安交通大学学报(医学版) 2014年3期
单 虎,魏 瑾,张 明,林 琳,闫 蕊,张 蓉
(西安交通大学医学院第二附属医院心内科,陕西西安 710004)
心肌细胞肥大是心脏在病理条件下维持正常功能的有效代偿机制,然而心肌细胞肥大失代偿可导致扩张性心肌病、心力衰竭和猝死的发生[1-2]。线粒体功能异常是参与心肌细胞肥大及失代偿过程中的重要机制[3],主要表现为线粒体膜电位降低,数目减少,聚集伴肿胀[4],线粒体DNA突变增多[5]以及线粒体内活性氧水平升高[6]。然而,肥大刺激[血管紧张素Ⅱ(AngⅡ)、内皮素-1、胰岛素样生长因子-1等]导致线粒体功能异常的机制尚未明确。目前,研究已初步证实NADPH氧化酶、线粒体σ1受体及线粒体KATP通道等参与肥大刺激介导的线粒体损伤[7-8]。钙网蛋白(calreticulin, CRT)是一种主要分布于内质网的钙离子结合蛋白,除参与调节蛋白折叠、细胞钙稳态和细胞黏附外[9],CRT还可通过影响线粒体膜电位和钙稳态调节线粒体功能[10]。而且,在心肌细胞肥大过程中CRT表达水平明显升高[11]。但是,CRT诱导的线粒体功能异常是否参与心肌细胞肥大尚未证实。本研究拟采用AngⅡ构建大鼠心肌细胞肥大模型,观察模型细胞中CRT表达和线粒体功能的变化及AngⅡ受体阻断剂对它们的影响。
1 材料与方法
1.1材料新生SD大鼠(雌雄不拘)购自西安交通大学医学院实验动物中心;胰蛋白酶和Ⅱ型胶原酶(MP公司),5-溴脱氧尿核苷(Brdu,Sigma公司);DMEM/F12培养基(Thermo公司),胎牛血清(Gibco公司);α-横纹肌肌动蛋白单克隆抗体(Santa Cruz公司),CRT单克隆抗体(CST公司),Alexa Fluor 594标记二抗、罗丹明123(Invitrogen公司),Hoechst 33258(上海碧云天);AngⅡ(Sigma公司),酶活性检测试剂盒(南京建成);Tris-Carb系液闪仪(Perkinelmer公司)、Hoefer转膜仪、实时定量PCR仪(Bio-Rad公司),荧光酶标仪、分光光度仪(上海天普)、倒置相差显微镜(Olympus公司)。常规试剂均为国产分析纯。
1.2新生大鼠心肌细胞原代分离培养与鉴定5只1 d龄SD大鼠消毒后移入超净台,无菌条件下迅速取出心脏,将左心室部分剪碎后移入1 g/L胰蛋白酶与0.5 g/L Ⅱ型胶原酶混合消化酶中,在37 ℃恒温摇床中消化7 min后吸取上清液与等体积含100 mL/L血清的完全培养基混合,重复消化8~10次。将所收集上清离心后以含100 mL/L血清的完全培养基(含100 U/mL青链霉素及0.1 mmol/L Brdu)重悬后置于37 ℃细胞培养箱内培养90 min,采用差速贴壁法去除贴壁的非心肌细胞。取培养第4天的心肌细胞常规固定通透后,以1∶200稀释α-横纹肌肌动蛋白单克隆抗体4 ℃孵育过夜,1∶200稀释的Alexa Fluor 594标记二抗室温孵育2 h,Hoechst 33258标记细胞核,荧光显微镜下观察。
1.3实验分组将同期分离培养的心肌细胞随机分为5组:模型组,共3组,分别以10-8mmol/L、10-7mmol/L、10-6mmol/L终浓度的AngⅡ干预48 h;缬沙坦组以10-6mmol/L终浓度缬沙坦预处理0.5 h后,再加入10-7mmol/L终浓度的AngⅡ继续干预48 h;空白对照组不予任何处理。
1.4心肌细胞表面积的测定各组心肌细胞干预结束后,于倒置相差显微镜下观察心肌细胞形态,每组随机挑选10个视野,各视野下随机挑选10个细胞,于40倍物镜下摄片。用Image-Pro Plus专业图像分析软件测量细胞表面积,各组细胞表面积用该组100个细胞表面积的平均值代表。
1.5蛋白质合成速率的测定采用3H-亮氨酸掺入法,各组心肌细胞干预结束后,PBS冲洗2遍,用含0.2 g/L EDTA的2.5 g/L胰酶将细胞消化为单细胞悬液,离心后以含1 μCi/mL3H-亮氨酸的PBS 1 mL重悬细胞,37 ℃水浴1.5 h后将心肌细胞转移至玻璃纤维滤膜上,以100 g/L三氯乙酸固定滤膜并烘干,将烘干后的滤膜放入含5 mL闪烁液的测定瓶中,用液体闪烁仪测定滤膜放射性。
1.6线粒体膜电位的测定将罗丹明123配制成5 μg/μL的母液,用PBS漂洗贴壁生长的心肌细胞3次,再各孔中加入含5 μg/mL终浓度的新鲜培养液,放入孵育箱中继续培养30 min,吸弃培养液,PBS轻轻洗涤细胞3遍,于荧光酶标仪检测荧光强度,激发波长488 nm,发射波长分别为535 nm和590 nm,红色与绿色荧光强度比值反映膜电位水平。
1.7酶活性测定按照酶活性检测试剂盒说明书操作,于分光光度仪550 nm和600 nm波长分别检测COX和SDH活性。
1.8RT-PCR检测使用Trizol试剂提取细胞总RNA,随后用RT-PCR试剂盒(TaKaRa)进行反转录,按照SYBRExScriptTMRT-PCR试剂盒说明在iQ5多色实时定量PCR检测系统上进行操作。CRT引物设计为5′-TTCTTGGACGGAGATGC-3′(正义),5′-CATCTTGGCTTGTCTGC-3′(反义);GAPDH,5′-TTGTGATGGGTGTGAACC-3′(正义),5′-TTCTGAGTGGCAGTGATG-3′(反义)。以NADPH为内部对照,采用2-ΔΔCt相对定量法计算各组中CRT基因表达。
1.9Westernblot检测使用RIPA裂解液提取细胞总蛋白,Bradford法测定蛋白质浓度,分别取300 μg蛋白质进行聚丙烯酰胺凝胶电泳,继而转印到PVDF膜,常规封闭后分别用CRT单克隆抗体(1∶1 000)及β-actin单克隆抗体(1∶3 000)4 ℃孵育过夜,PBST洗涤后以相应二抗室温孵育2 h,ECL法显影并于暗室内曝光。以β-actin作为内部对照,采用Image-Pro Plus图像分析软件分析各蛋白条带的积分吸光度值。
1.10统计学方法采用SPSS 18.0统计软件分析,数据以均数±标准差表示,两组间的比较采用t检验,以P<0.05为差异有统计学意义。
2 结 果
2.1原代培养乳鼠心肌细胞的纯度鉴定在培养的心肌细胞中以α-横纹肌肌动蛋白单克隆抗体为一抗,经免疫荧光细胞化学鉴定,心肌细胞纯度在90%以上(图1、表1)。
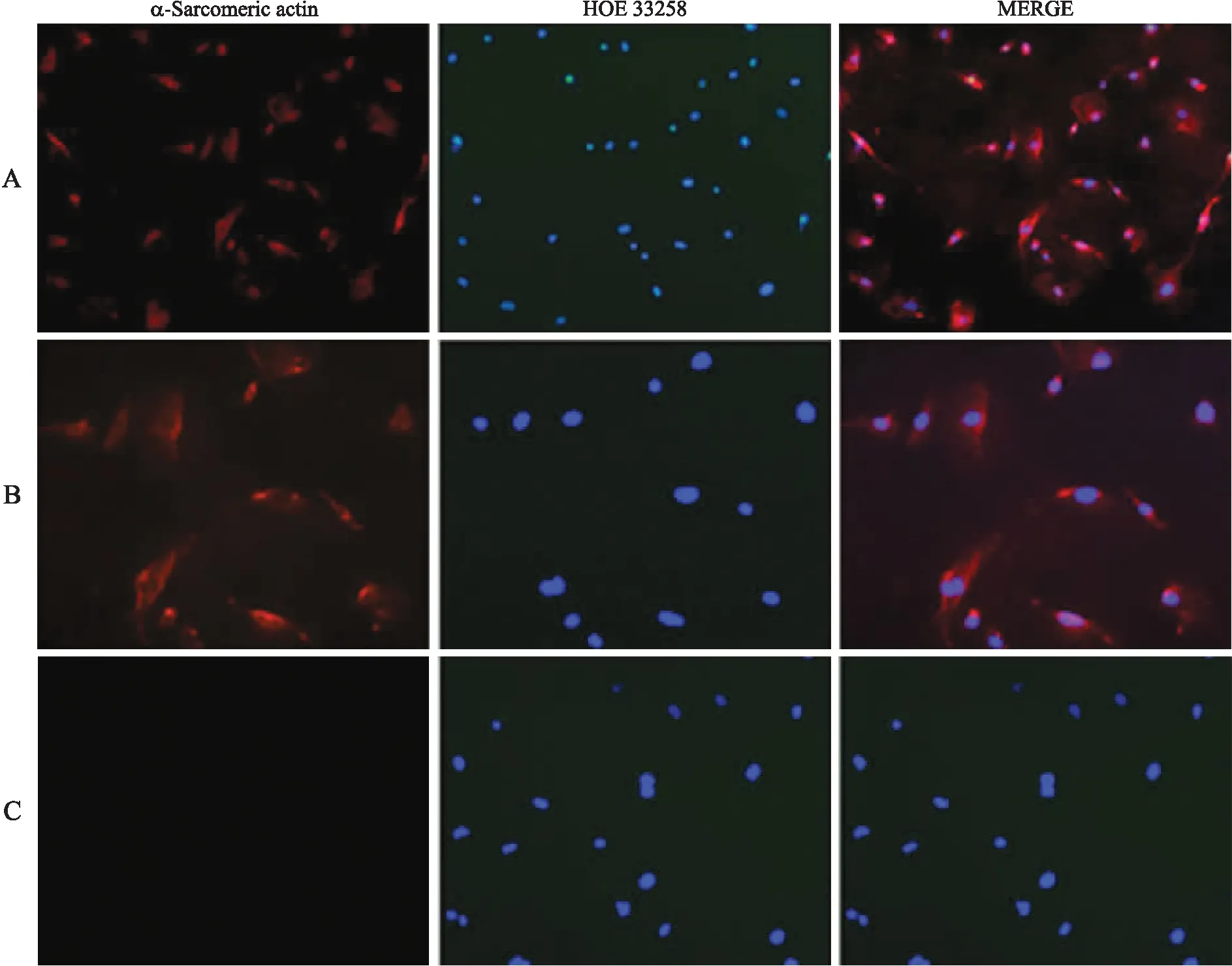
图1原代培养乳鼠心肌细胞的纯度鉴定(免疫细胞化学染色)
Fig.1 The identification of neonatal rat cardiomyocytes (immunocytochemistry)
A:实验组(×40);B:实验组(×100);C:阴性对照组(PBS代替一抗,×40)。
表1以α-横纹肌肌动蛋白的阳性率评定原代培养乳鼠心肌细胞的纯度
Tab.1 The purity of neonatal rat cardiomyocytes identified by α-sarcomeric actin positive rate
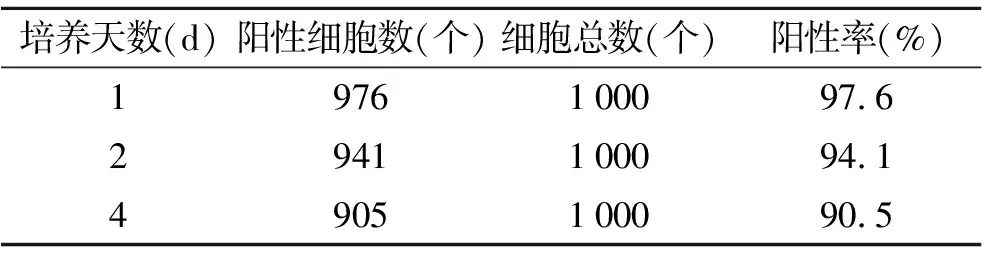
培养天数(d)阳性细胞数(个)细胞总数(个)阳性率(%)1976100097.62941100094.14905100090.5
2.2缬沙坦对AngⅡ诱导心肌细胞肥大的影响分别用3种不同浓度AngⅡ干预的模型组心肌细胞表面积、蛋白质合成速率明显高于对照组(P<0.01),提示心肌细胞肥大模型构建成功。与3个模型组相比,缬沙坦组心肌细胞表面积、蛋白质合成速率均明显降低(P<0.05,图2)。
2.3缬沙坦对AngⅡ诱导心肌细胞线粒体功能异常的影响与对照组相比,3个模型组线粒体膜电位、COX及SDH活性均明显降低,而与3个对照组相比,缬沙坦预处理可有效抑制AngⅡ诱导的线粒体膜电位和酶活性下降(P<0.05,图3)。
2.4缬沙坦对AngⅡ诱导心肌细胞肥大中CRT表达的影响与对照组相比,模型组心肌细胞中CRT mRNA及蛋白水平随着AngⅡ浓度增大而逐渐升高,而与3个模型组相比,缬沙坦预处理可明显抑制CRT表达上调(P<0.01,图4)。
3 讨 论
CRT在调节心肌线粒体功能中的作用已经得到广泛的认同,而其是否在心肌肥大发生过程中参与线粒体损伤尚未得到证实。本研究通过体外实验观察了心肌肥大发生过程中CRT表达的变化及缬沙坦对其表达的影响。
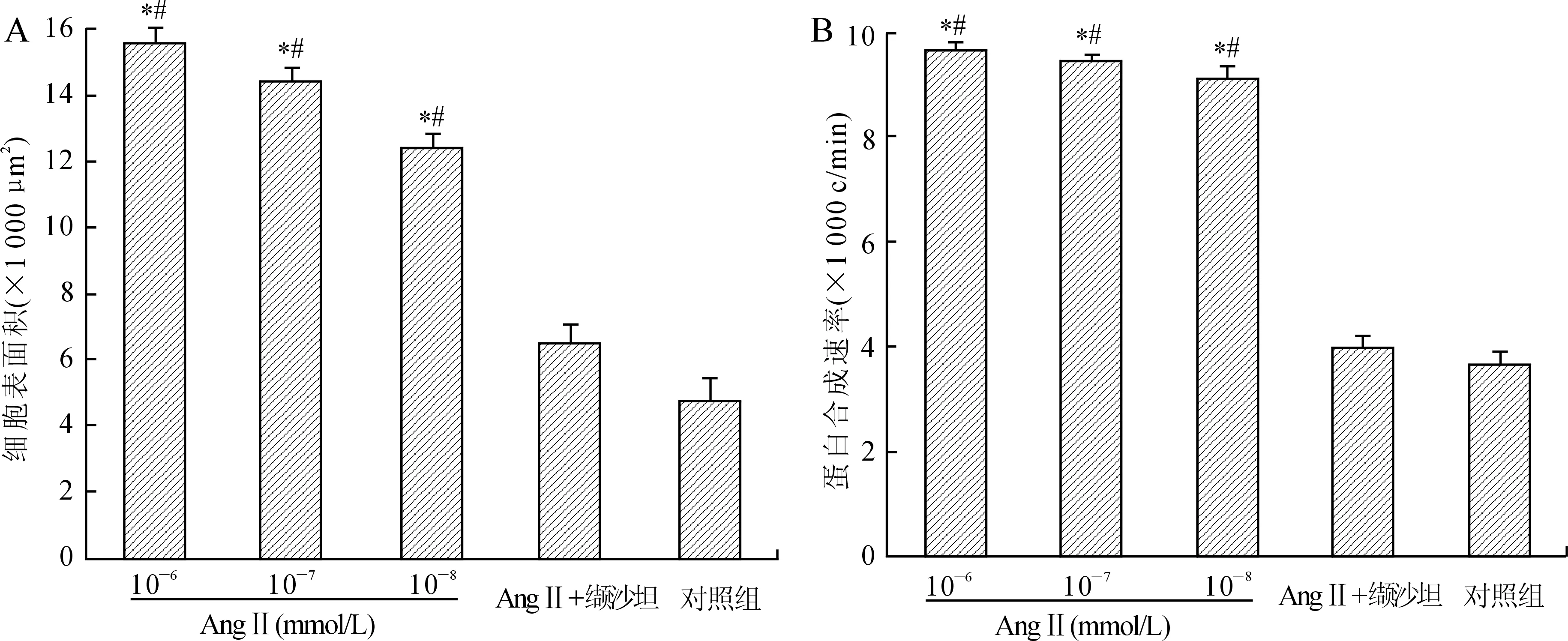
图2各组细胞表面积、蛋白质合成速率的比较
Fig.2 Comparison of cell surface area and protein synthesis rate between the groups
A:各组细胞表面积;B:各组细胞蛋白合成速率。与对照组相比,*P<0.01;与缬沙坦组相比,#P<0.01。
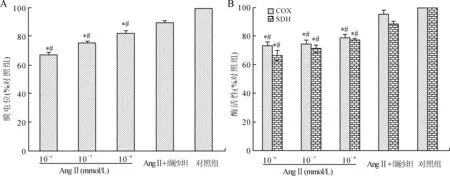
图3各组细胞线粒体膜电位、呼吸链酶活性的比较
Fig.3 Comparison of mitochondrial membrane potential level and enzyme activities between the groups
A:各组细胞线粒体膜电位;B:各组细胞线粒体中呼吸链酶活性。与对照组相比,*P<0.05;与缬沙坦组相比,#P<0.05。

图4各组细胞CRT表达水平的比较
Fig.4 Comparison of CRT expression between the groups
A:各组细胞CRT mRNA相对水平;B:各组细胞CRT蛋白相对水平。与对照组相比,*P<0.01;与缬沙坦组相比,#P<0.01。
在我们构建的心肌肥大细胞模型中,3个浓度梯度的AngⅡ均可诱导原代心肌细胞出现细胞表面积增大、蛋白合成速率加快等肥大特征及线粒体膜电位、酶活性降低等线粒体功能受损表现,同时CRT表达水平随AngⅡ浓度增加呈现剂量依赖性升高,而缬沙坦预处理可消除AngⅡ诱导的CRT表达上调作用,并减轻AngⅡ诱导的线粒体功能损伤。结合既往研究所证实CRT表达上调可导致线粒体功能受损,上述实验能够初步证实:CRT介导的线粒体损伤可能参与心肌肥大过程。
CRT是一种主要分布在内质网中的钙结合蛋白,具有调节细胞内钙稳态、钙离子相关信号通路、基因表达和细胞黏附的生物学作用[12]。CRT在胚胎期心脏中表达较高,是心脏发育必不可少的重要因素[13]。然而,出生后心脏中CRT表达水平迅速下降,当CRT过度表达时则会出现心律失常、心腔扩大、心源性猝死等严重后果[14-15]。目前,CRT在心血管疾病发病机制中的作用得到越来越多研究者的重视,ARNAUDEAU等[10]已发现CRT介导线粒体钙稳态失衡是心肌细胞功能异常的重要机制。本课题组前期以扩张性心肌病大鼠模型为研究对象,发现其心肌组织中CRT表达升高,并伴随信号转导和转录激活子3(STAT3)磷酸化过程受阻,线粒体损伤发生;离体培养大鼠心肌细胞CRT表达下调后,STAT3磷酸化受阻及线粒体损伤得到部分逆转。由此可见,CRT通过CRT-STAT3信号通路介导的线粒体损伤参与扩张性心肌病的发病过程[16]。早在1997年,TSUTSUI等人就已经发现压力负荷诱导的心肌肥大模型中CRT表达异常升高,但其在心肌肥大过程中的具体作用及机制尚未明确。本研究发现心肌肥大细胞模型中CRT表达水平升高,并呈现AngⅡ浓度依赖性,同时,心肌细胞线粒体功能下降,而给予缬沙坦预处理后,AngⅡ不能诱导CRT表达升高,随着CRT表达水平的降低,线粒体功能也得到一定程度的恢复。因此,本实验结果可初步证实CRT介导的线粒体损伤参与AngⅡ诱导心肌肥大过程。
综上所述,在AngⅡ刺激下,CRT表达升高所诱导的线粒体功能异常可能是心肌肥大的重要机制之一。该研究有助于阐述心肌肥大过程中线粒体功能受损的可能机制,为进一步的基础与临床研究奠定了理论基础。
参考文献:
[1] 徐浩,孙丽君,张建保,等. 钙依赖途径在高糖致心肌细胞肥大中的作用[J]. 西安交通大学学报:医学版,2012,33(2):156-160.
[2] 周娟,徐心,刘进军,等. AT2对成年压力超负荷性肥大心肌细胞TNFα、IL-1β及IL-6分泌的调控[J]. 西安交通大学学报:医学版,2008,29(1):25-28.
[3] FREY N, OLSON EN. Cardiac hypertrophy: the good, the bad, and the ugly[J]. Annu Rev Physiol, 2003, 65(1):45-79.
[4] FINCKENBERG P, ERIKSSON O, BAUMANN M, et al. Caloric restriction ameliorates angiotensin II-induced mitochondrial remodeling and cardiac hypertrophy[J]. Hypertension, 2012, 59(1):76-84.
[5] KIM T, THU VT, HAN IY, et al. Does strong hypertrophic condition induce fast mitochondrial DNA mutation of rabbit heart?[J]. Mitochondrion, 2008, 8(3):279-283.
[6] DAI DF, JOHNSON SC, VILLARIN JJ, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Gαq overexpression-induced heart failure[J]. Circ Res, 2011, 108(7):837-846.
[7] ZHANG GX, LU XM, KIMURA S, et al. Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation[J]. Cardiovas Res, 2007, 76(2):204-212.
[8] TAGASHIRA H, ZHANG C, LU YM, et al. Stimulation of σ1-receptor restores abnormal mitochondrial Ca2+mobilization and ATP production following cardiac hypertrophy[J]. Biochim Biophys Acta, 2013, 1830(4):3082-3094.
[9] GOLD LI, EGGLETON P, SWEETWYNE MT, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and disease[J]. FASEB J, 2010, 24(3):665-683.
[10] ARNAUDEAU S, FRIEDEN M, NAKAMURA K, et al. Calreticulin differentially modulates calcium uptake and release in the endoplasmic reticulum and mitochondria[J]. J Biol Chem, 2002, 277(48):46696-46705.
[11] TSUTSUI H, ISHIBASHI Y, IMANAKA-YOSHIDA K, et al. Alterations in sarcoplasmic reticulum calcium-storing proteins in pressure-overload cardiac hypertrophy [J]. Am J Physiol-Heart C, 1997, 272(1):H168-H175.
[12] GELEBART P, OPAS M, MICHALAK M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum[J]. Int J Biochem Cell B, 2005, 37(2):260-266.
[13] MESAELI N, NAKAMURA K, ZVARITCH E, et al. Calreticulin is essential for cardiac development[J]. J Cell Biol, 1999, 144(5):857-868.
[14] HATTORI K, NAKAMURA K, HISATOMI Y, et al. Arrhythmia induced by spatiotemporal overexpression of calreticulin in the heart[J]. Mol Genet Metab, 2007, 91(3):285-293.
[15] NAKAMURA K, ROBERTSON M, LIU G, et al. Complete heart block and sudden death in mice overexpressing calreticulin[J]. J Clin Invest, 2001, 107(10):1245-1253.
[16] ZHANG M, WEI J, SHAN H, et al. Calreticulin-STAT3 signaling pathway modulates mitochondrial function in a rat model of furazolidone-induced dilated cardiomyopathy[J]. PLoS One, 2013, 8(6):e66779.
猜你喜欢
发明与创新(2023年30期)2023-10-11 01:37:12
小学生学习指导(高年级)(2023年3期)2023-03-31 06:03:22
小学生学习指导(高年级)(2022年3期)2022-03-29 07:49:16
安家(校外教育)(2022年6期)2022-01-03 11:47:06
世界科学技术-中医药现代化(2021年7期)2021-11-04 08:10:42
小学生导刊(高年级)(2017年2期)2017-06-10 02:40:42
湖北理工学院学报(2015年1期)2015-02-27 15:02:36
卫生职业教育(2014年16期)2014-05-16 03:48:28
中国药理学通报(2014年2期)2014-05-09 08:22:33
中医研究(2014年2期)2014-03-11 20:28:18
