
结核分枝杆菌Hsp16.3表达与感染小鼠肺泡巨噬细胞凋亡的关系
2014-06-27 10:36:04庹清章董江涛田玺择刘云霞董伟杰刘丹霞张万江
西安交通大学学报(医学版) 2014年3期
庹清章,董江涛,田玺择,刘云霞,董伟杰,刘丹霞,李 微,吴 芳,章 乐,张万江
(石河子大学医学院病理生理学教研室,新疆地方与民族高发病教育部重点实验室,新疆石河子 832002)
结核病是由结核分枝杆菌(MycobacteriumTuberculosis, MTB)引起的一种世界性传染病,也是单一致病菌感染导致死亡率最高的感染性疾病。当机体吸入含有MTB的悬浮颗粒时,MTB先感染肺泡巨噬细胞[1]。一般认为,结核病的发生、发展过程中伴随着机体免疫细胞的凋亡。宿主巨噬细胞凋亡可杀死寄生于其内的MTB,阻止MTB在体内播散,而未被清除的MTB仍潜伏于宿主巨噬细胞中。近期研究发现,MTB小分子热休克蛋白(small heat shock proteins, sHsps)Hsp16.3对MTB在宿主巨噬细胞中的潜伏有重要作用。本文将通过对MTB国际标准强毒株H37Rv菌株(H37Rv)与MTB国际标准强毒株H37Rv株Hsp16.3基因缺失突变菌株(△H37Rv)的对比研究,探讨MTB小分子热休克蛋白Hsp16.3的表达与宿主巨噬细胞凋亡的关系及机制,以期为结核病的治疗带来新的希望。
1 材料与方法
1.1模型建立和分组选用8周龄雄性SPF级(无特定病原体动物)昆明小鼠120只(购于石河子大学实验动物中心),体质量18~20 g,随机分为△H37Rv(本课题组制备)感染组、H37Rv(中国食品药品检定研究院)感染组和无菌生理盐水对照组(对照组)。每组40只,1、3、5、7、9、11、13、15 d各设5只。方法参见文献[2]:在生物安全柜内,取在改良罗氏培养基上生长2~3周状态良好的MTB△H37Rv菌落,置灭菌研菌器中,加少量含0.5 mL/L Tween-20的生理盐水溶液充分研磨,使其成均匀浑浊的菌悬液。麦氏比浊法调细菌密度约1.0×107cfu/mL,于每只小鼠尾静脉内注射0.3 mL(约含活菌量3×106cfu/mL)。H37Rv组也按上述方法建立。对照组仅注射灭菌生理盐水溶液。感染小鼠置生物安全三级实验室内,IVC笼具中饲养。
1.2小鼠肺泡巨噬细胞的分离小鼠于感染后第1、3、5、7、9、11、13、15 d经眼球放血,脱颈处死,暴露气管,用消毒好的组织剪在气管上做一切口(切勿剪断),用连有注射器的无菌软皮管从切口处插入气管,丝线固定,用预热至37 ℃的PBS液行支气管肺泡灌洗,1 mL×10次,收集支气管肺泡灌洗液,4 ℃ 1 500 r/min离心10 min,PBS液洗涤细胞1次。弃上清,加入含100 mL/L胎牛血清的DMEM培养液,转入细胞培养瓶中。置37 ℃、50 mL/L CO2孵箱中培养4 h,弃上清液和非贴壁的细胞,贴壁的即为小鼠肺泡巨噬细胞。
1.3激光共聚焦显微镜观察各组感染小鼠肺泡巨噬细胞收集感染小鼠肺泡巨噬细胞,经爬片、固定、封闭后,滴加稀释后的一抗(稀释度1∶1 500),均匀铺于玻片上,湿盒内放置,4 ℃过夜。PBS冲洗3 min×3次,滴加荧光二抗(稀释度1∶500),室温2 h,PBS冲洗干净,硝酸甘油封片,于激光共聚焦显微镜下观察荧光强度及着色部位。
1.4小鼠肺泡巨噬细胞凋亡检测收集各组、各时间点小鼠肺泡巨噬细胞,Annexin V-FITC/PI染色,1 h内流式细胞仪检测细胞凋亡,Cellquest软件获取数据。
1.5Westernblot检测收集各组各时间点巨噬细胞,加细胞裂解液冰上裂解30 min,低温离心12 000r/min×5 min×2次,上清液为提取总蛋白。测定蛋白浓度。进行SDS-PAGE凝胶(120 g/L分离胶+50 g/L浓缩胶)电泳及半干转膜(恒压23V、维持45 min)至聚偏二氟乙烯(PVDF)膜上,50 g/L的脱脂奶粉封闭1.5 h,TBST洗脱液洗脱5 min×3次。加稀释一抗(均为兔多克隆抗体,Caspase-3为1∶1 000, Bcl-2为1∶300,β-actin以1∶1 000),4 ℃孵育过夜。TBST洗脱液洗膜5 min×3次。加入HRP标记的相应二抗作用液(1∶3 000稀释),室温孵育2 h, 用ECL发光液显影。以Quantity One成像系统采集图像,将获取的图像用ImageJ图像处理软件分析蛋白相对表达量。
1.6统计学分析使用SPSS 13.0统计软件,多组间比较采用单因素方差分析和LSD-t检验,以P<0.05为差异有统计学意义。
2 结 果
2.1激光共聚焦显微镜观察各组感染小鼠肺泡巨噬细胞感染小鼠肺泡巨噬细胞内MTB膜上的热休克蛋白Hsp65与一抗(只针对MTB的65 ku的小鼠单克隆抗体)结合,再与绿色荧光(FITC)标记的二抗结合。在激光共聚焦显微镜下,MTB H37Rv菌株和△H37Rv感染组小鼠肺泡巨噬细胞内均可见大量绿色荧光,仅对照组未见绿色荧光(图1),表明MTBH37Rv菌株和△H37Rv菌株感染小鼠肺泡巨噬细胞的动物模型建立成功。
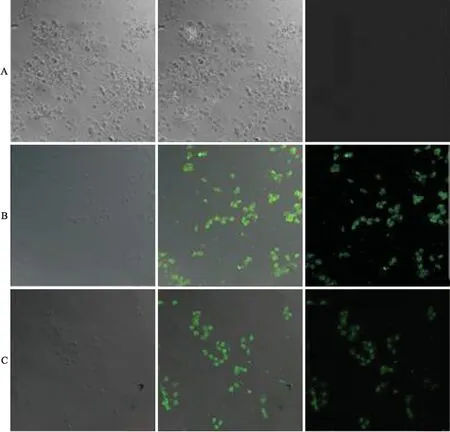
图1激光共聚焦显微镜观察各组小鼠肺泡巨噬细胞
Fig.1 Alveolar macrophages of infected mice under CLSM (×200)
A:对照组;B:H37Rv组;C:△H37Rv组。
2.2各组小鼠肺泡巨噬细胞的凋亡情况经流式细胞技术检测,Cellquest软件分析,可见各组细胞凋亡率(图2)并建立了各组小鼠肺泡巨噬细胞的凋亡率-时间曲线(图3)。△H37Rv组巨噬细胞的凋亡率逐渐上升,至感染7 d时达到高峰,随后逐渐降低。1~7 d内,各时间点△H37Rv组巨噬细胞凋亡率均显著高于H37Rv组,差异有统计学意义(P<0.05),随时间延长发生了逆转,△H37Rv组巨噬细胞凋亡率9~11 d内低于H37Rv组,而13~15 d内高于H37Rv菌株组,差异均有统计学意义(P<0.05)。

图2流式细胞仪检测各组小鼠的肺泡巨噬细胞的凋亡情况
Fig.2 Apoptosis of alveolar macrophages of infected mice detected by flow cytometry
A:对照组;B:H37Rv组;C:△H37Rv组。
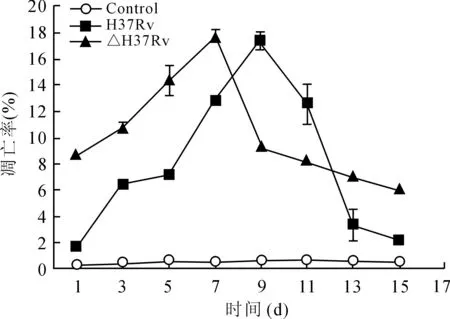
图3H37Rv组、△H37Rv组和对照组巨噬细胞凋亡率-时间曲线
Fig.3 Relationship between apoptosis rate and infection time of the microphages in the three groups
2.3各组小鼠肺泡巨噬细胞内Caspase-3和Bcl-2蛋白的表达情况Western Blot检测结果显示各时间点H37Rv组和△H37Rv组均可诱导巨噬细胞Caspase-3蛋白表达,与对照组相比,差异均有统计学意义(P<0.05);随时间延长,H37Rv和△H37Rv组巨噬细胞Caspase-3的表达水平亦相应增加,至感染第7天时达到高峰,而且△H37Rv组在13 d时出现第二次高峰;△H37Rv组Caspase-3表达水平始终高于H37Rv组(图4、图5)。
H37Rv组和△H37Rv组各时间点均可诱导巨噬细胞Bcl-2蛋白表达,与对照组相比,差异均有统计学意义(P<0.05)。△H37Rv组巨噬细胞Bcl-2蛋白的表达水平在感染1~7 d无明显变化,9 d以后逐渐升高,但表达水平始终低于H37Rv组,且7 d后更为显著(图4、图6)。
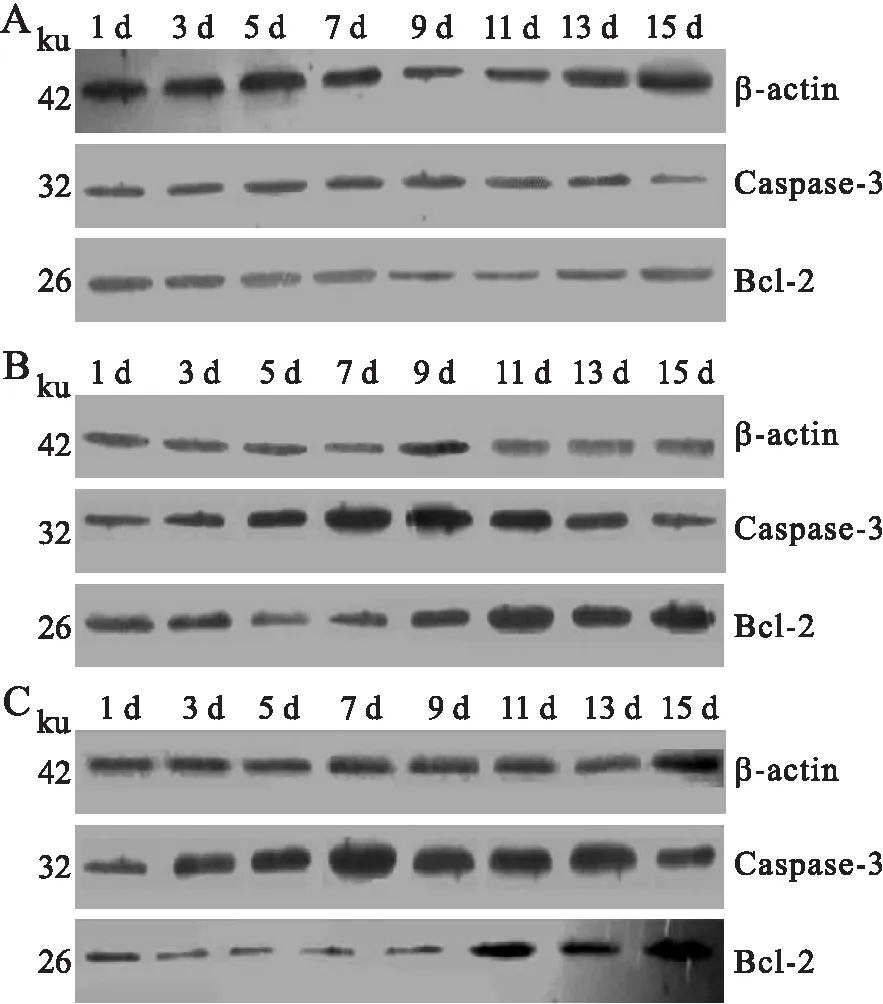
图4Westernblot检测巨噬细胞内Caspase-3及Bcl-2蛋白表达情况
Fig.4 Western blot analysis of the protein expressions of Caspase-3 and Bcl-2 in the macrophages
A:对照组;B:H37Rv组;C:△H37Rv组。
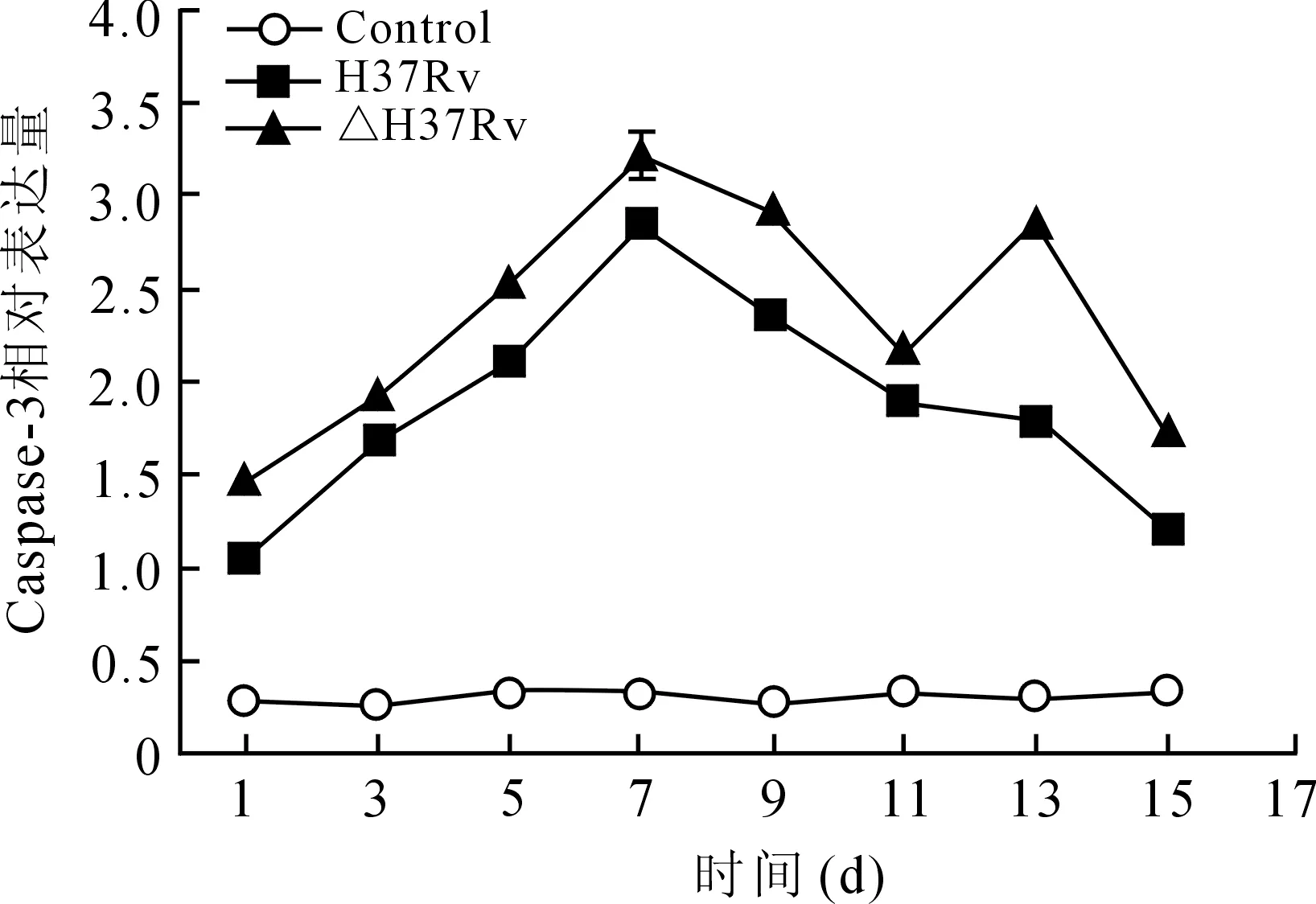
图5各组巨噬细胞Caspase-3的相对表达量-时间曲线
Fig.5 Relationship between relative expression of Caspase-3 and infection time of the microphages in the three groups

图6各组巨噬细胞Bcl-2的相对表达量-时间曲线
Fig.6 Relationship between relative expression of Bcl-2 and infection time of the microphages in the three groups
3 讨 论
MTB是典型的胞内致病菌,主要在巨噬细胞等宿主免疫系统细胞内存活和繁殖。MTB感染人体后,主要被宿主巨噬细胞吞噬,未被机体免疫系统清除而潜伏下来的MTB也主要寄生于宿主巨噬细胞内[3]。MTB感染的后果以及结核病的发生与否与宿主巨噬细胞密切相关[4]。宿主巨噬细胞发生凋亡后,可杀死寄生于其内的MTB,阻止MTB在体内播散,并能激活邻近未感染的巨噬细胞,增强机体对MTB的杀伤能力[5]。因此, 干预和调控宿主巨噬细胞的凋亡进程,可促进MTB在宿主巨噬细胞中的成功存活[6]。
在MTB感染过程中,MTB与宿主巨噬细胞相互作用和适应,调控宿主巨噬细胞凋亡,MTB有诱导或抑制巨噬细胞凋亡的作用,当这两种作用达到平衡时,MTB则以休眠状态存在于巨噬细胞中,形成所谓的“逃避”,使自身的存活、繁殖与宿主的一系列免疫反应达到一种动态平衡[7]。
Hsp16.3是近年发现的MTB中一个存在于膜上的主要抗原蛋白,由144个氨基酸组成,分子质量为16 277 u,等电点为4.85,序列分析表明属于sHsps家族。MTB小分子热休克蛋白Hsp16.3是一个组成性表达蛋白,正常条件下有少量表达[8],在MTB进入静止生长期时显著表达[9-10]。MTB进入巨噬细胞后大量表达合成MTB小分子热休克蛋白Hsp16.3;敲除Hsp16.3基因而得到的突变菌株在体外的生长情况与正常菌株相同,但突变菌株不能在小鼠骨髓衍生的巨噬细胞以及THP-1细胞株中生长[11-12],说明MTB小分子热休克蛋白Hsp16.3可能对MTB在巨噬细胞中的潜伏起着保护作用。同时有研究进一步证明,MTB进入宿主巨噬细胞时诱导大量表达的Hsp16.3对MTB能够在宿主巨噬细胞内长期生长、繁殖和致病有重要作用,有助于MTB细胞膜增厚以及增强MTB抵抗胁迫环境,如NO、氧自由基、缺氧等因素的抗氧化能力等,同时使MTB在宿主巨噬细胞内转变成静止期,有助于MTB在宿主巨噬细胞内成为滞留菌,长期在宿主巨噬细胞内生长繁殖[13-14]。小鼠体内实验研究表明,敲除Hsp16.3基因而得到的突变菌株在感染早期和晚期对小鼠肺泡巨噬细胞有更强的致凋亡作用,这进一步说明MTB小分子热休克蛋白Hsp16.3的表达抑制了小鼠肺泡巨噬细胞的凋亡[15]。尽管发现MTB小分子热休克蛋白Hsp16.3对于MTB在宿主巨噬细胞内的生存是必需的,但是其在体内的生理功能以及相关的保护机制却并不清楚[16]。
天冬氨酸特异性半胱氨酸蛋白酶(cysteine aspartate-specific protease, Caspase)在细胞凋亡的过程中起关键作用,细胞凋亡各个阶段都能检测到不同程度的Caspase增高。Caspase-3是凋亡过程中最关键的蛋白酶,是多种死亡受体介导的凋亡途径的共同下游效应部分。本研究显示,在感染的早期(1~7 d)和晚期(13~15 d),△H37Rv组巨噬细胞内的Caspase-3表达水平高于H37Rv组;而在感染早期和晚期△H37Rv组巨噬细胞凋亡率也显著高于H37Rv组,这可能是Caspase-3蛋白表达量的增加促进了巨噬细胞的凋亡,表明MTB小分子热休克蛋白Hsp16.3的表达抑制了巨噬细胞凋亡的核心因子Caspase-3的表达,这可能是MTB小分子热休克蛋白Hsp16.3抑制巨噬细胞凋亡、保护MTB处于休眠状态的重要作用机制之一。
B细胞淋巴瘤/白血病-2家族位于线粒体外膜上,包括抗凋亡蛋白(Bcl-2、Bcl-XL等)和促凋亡蛋白(Bax、Bad等)。其中Bcl-2是一种重要的凋亡抑制蛋白,具有保护细胞的功能,Bcl-2的过度表达可引起细胞核谷胱甘肽的积聚,导致核内氧化还原平衡改变,从而降低了Caspase的活性,阻止凋亡诱导因子和细胞色素C从线粒体释放,抑制凋亡[17]。本研究结果显示,在感染早期(1~7 d),△H37Rv感染组Bcl-2的表达水平无明显变化,但在9 d后逐渐升高,而巨噬细胞凋亡率却在此时逐渐下降,但△H37Rv感染组Bcl-2的表达水平始终低于H37Rv组且7 d后表现更为显著,这表明Bcl-2表达增加对巨噬细胞的凋亡形成有明显抑制作用,而MTB小分子热休克蛋白Hsp16.3的表达促进了Bcl-2蛋白的表达。
由此推断:在MTB感染的早期和晚期,MTB小分子热休克蛋白Hsp16.3的表达能有效抑制小鼠肺泡巨噬细胞凋亡,可能通过抑制Caspase-3表达以及促进Bcl-2表达而实现。这将为结核病的预防、控制和根除提供新的靶点,同时也为结核病治疗时间的选择提供了理论依据。
参考文献:
[1] 庹清章,张万江.结核分枝杆菌感染中巨噬细胞凋亡的研究进展[J]. 中国人兽共患病学报,2012,28(5):496-499.
[2] 董江涛,徐芳,田玺择,等. 不同毒力结核分枝杆菌感染小鼠对肺泡巨噬细胞的凋亡率及其时相性变化的影响[J]. 中国免疫学杂志,2012,28(5):389-392.
[3] 刘云霞,张万江. 结核分枝杆菌与宿主巨噬细胞相互作用的研究进展[J]. 中国细胞生物学学报,2012,34(6):617-622.
[4] DANELISHVILI L, MCGARVEY J, LI YJ, et al. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells[J]. Cell Microbiol, 2003, 5(9):649-660.
[5] RIENDEAU CJ, KORNFELD H. THP-1 cell apoptosis in response to Mycobacterial infection[J]. Infect Immun, 2003, 71(1):254-259.
[6] KEANE J, REMOLD HG, KORNFELD H. Virulent mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages[J]. J Immunol, 2000, 164(4):2016-2020.
[7] SLY LM, HINGLEY-WILSON SM, REINER NE, et al. Survival of mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1[J]. J Immunol, 2003, 170(1):430-437.
[8] MITRA G, SAHA A, GUPTA TD, et al. Chaperone-mediated inhibition of tubulin self-assembly[J]. Proteins, 2007, 67(1):112-120.
[9] FU X, CHANG Z. Identification of bis-ANS binding sites in Mycobacterium tuberculosis small heat shock protein Hsp16.3: evidences for a two-step substrate-binding mechanism[J]. Biochem Biophys Res Commun, 2006, 349(1):167-171.
[10] FU X, CHANG Z. Identification of a highly conserved progly doublet in non-animal small heat shock proteins and characterization of its structural and functional roles in Mycobacterium tuberculosis Hsp16.3[J]. Biochemistry(Moscow), 2006, 71 (Suppl 1):S83-S90.
[11] FU X, ZHANG H, ZHANG X, et al. A dual role for the N-terminal region of Mycobacterium tuberculosis Hsp16.3 in self-oligomerization and binding denaturing substrate proteins[J].J Biol Chem, 2005, 280(8):6337-6348.
[12] PRENETA R, PAPAVINASASUNDARAM KG, COZZONE AJ, et al. Autophosphorylation of the 16 kDa and 70 kDa antigens (Hsp 16.3 and Hsp 70) of Mycobacterium tuberculosis[J]. Microbiology, 2004, 150(Pt 7):2135-2141.
[13] FU X, CHANG Z. Temperature-dependent subunit exchange and chaperone-like activities of Hsp16.3, a small heat shock protein from Mycobacterium tuberculosis[J]. Biochem Biophys Res Commun, 2004, 316(2):291-299.
[14] FU X, LIU C, LIU Y, et al. Small heat shock protein Hsp16.3 modulates its chaperone activity by adjusting the rate of oligomeric dissociation[J]. Biochem Biophys Res Commun, 2003, 310(2):412-420.
[15] 庹清章,董江涛,田玺择,等. 结核分枝杆菌小分子热休克蛋白Hsp16.3基因缺失突变菌株对感染小鼠肺泡巨噬细胞凋亡率的影响及其时相性变化[J]. 石河子大学学报:自然科学版,2013,31(1):54-60.
[16] VALDEZ MM, CLARK JI, WU GJ, et al. Functional similarities between the small heat shock proteins Mycobacterium tuberculosis HSP 16.3 and human alphaB-crystallin[J]. Eur J Biochem, 2002, 269(7):1806-1813.
[17] GIRIS M, ERBIL Y, DEPBOYLU B, et al. Heme oxygenase-1 prevents hyperthyroidism induced hepatic damage via an antioxidant and antiapoptotic pathway[J]. J Surg Res, 2010, 164(2):266-275.
猜你喜欢
现代临床医学(2021年4期)2021-07-31 07:55:44
科学(2020年3期)2020-11-26 08:18:22
当代水产(2020年3期)2020-06-15 12:03:02
家庭医学(下半月)(2020年4期)2020-05-30 12:42:40
国际呼吸杂志(2019年8期)2019-04-29 09:15:12
国际呼吸杂志(2019年8期)2019-04-29 09:15:04
老年医学与保健(2017年6期)2017-02-06 05:29:53
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:42
哈尔滨医药(2015年4期)2015-12-01 03:57:56
医学研究杂志(2015年12期)2015-06-10 06:57:46
