
基于高通量测序的寒地沼气池微生物群落解析
2014-06-15 17:14李树本
哈尔滨工业大学学报 2014年4期
赵 光,马 放,孙 婷,李树本,游 空,赵 贞
基于高通量测序的寒地沼气池微生物群落解析
赵 光1,2,马 放1,2,孙 婷3,李树本4,游 空4,赵 贞1,2
(1.哈尔滨工业大学城市水资源与水环境国家重点实验室,150090哈尔滨;2.哈尔滨工业大学市政环境工程学院,150090哈尔滨;3.辽宁工业大学化学与环境工程学院,121001辽宁锦州;4.海林农场,150000黑龙江牡丹江)
为实现寒地沼气发酵系统高效、稳定运行,应建立基于沼气发酵微生物群落的复合调控策略.本研究耦合454高通量测序和PCR-DGGE分析方法,对北方规模最大的海林农场沼气池内细菌及产甲烷古菌群落结构进行解析.取沼气池稳定运行期沼液样品,分析系统内微生物群落多样性.结果显示,共获得1 297条高质量微生物序列,在属和种的分类水平上,至少存在581个细菌属和666个细菌种.优势菌群有Firmicutes、Bacteroidetes及Proteobacteria,相对丰度分别为46.39%、21.41%和18.98%.优势属(相对丰度>5.0%)包括Proteiniphilum、Spirochaeta和Wolinella.DGGE分析结果表明,产甲烷古菌包括Methanocorpusculum sp.、Methanosaeta sp.、Methanobacterium sp.及Methanosarcina sp..,表明沼气池的产甲烷途径以乙酸代谢类型为主,水解、酸化过程主要由来自动物消化系统内的细菌完成.
沼气;微生物群落;海林农场;牛粪;454焦磷酸测序技术
应用厌氧生物技术处理农业废弃物产沼气,可以作为石化能源的替代能源,而且对于改善农村环境质量、调整能源消费结构等均具有重要意义[1-2].厌氧发酵产沼气是一系列复杂的生物学过程,在有机物降解的不同阶段都会由具有相应功能的微生物作用.这些微生物的代谢过程相互抑制,同时相互依存,共同维系厌氧发酵系统的稳定运行[3].有机物厌氧发酵的产甲烷过程中,非产甲烷菌群和产甲烷菌群既相互竞争,又相互依存.水解、产酸菌群为产甲烷菌群提供产甲烷底物基质,同时如果产酸代谢旺盛就会严重抑制产甲烷菌群的活性;产甲烷菌群也为水解、产酸菌群的正向代谢解除反馈抑制,并创造热力学上的优越条件.因此,调控厌氧消化系统内两大功能菌群的代谢平衡是获得稳定、高效产气率的关键.深入解析厌氧发酵系统微生物多样性和群落结构,对于厌氧发酵系统的升级改造及工艺优化均有重要指导作用.近年来,国内外学者基于传统的分子生物学技术,对厌氧发酵系统的微生物群落开展了大量研究,也发现了一些典型的参与厌氧发酵过程的微生物[4-6].但由于技术手段的局限性,无法对发酵过程微生物群落结构进行全面深层次的研究和解析.
高通量测序技术已广泛应用于各学科研究,拓宽了对于环境微生物研究的尺度与深度.目前,普遍应用的大规模高通量测序平台主要有Roche公司的454焦磷酸测序技术(454 pyrosequencing technology)、ABI公司的SOLiD(supported oligo ligation detetion)以及Illumina公司的Solexa等.此外,深入解析厌氧发酵系统运行过程中微生物群落结构的动态演替及功能的改变,对于研究系统运行不稳定机理及制定有效调控策略,使其实现高效、稳定运行具有重要意义[7].本研究以北方寒地最大的沼气发酵系统——黑龙江海林农场沼气池为研究对象,耦合454焦磷酸高通量测序和PCR-DGGE分析技术,解析其稳定运行时的微生物多样性和群落结构组成.以期为运行工艺的优化、系统升级改造、提高冬季甲烷转化效率以及制定高效的调控策略提供理论参考.
1 实 验
1.1 沼气池发酵原料及运行参数
海林农场沼气发酵系统生产模式如图1所示,沼气池总容积1 920 m3,由8个分隔池体组成.发酵原料为牛粪,主要来自农场圣澳奶牛养殖场.沼气池以半连续发酵工艺运行,干物质质量分数8%,发酵温度为35℃,内置机械搅拌系统,间歇式启动;有机负荷(organic loading rate,OLR)和水力停留时间(hydraulic retention time,HRT)分别为2 kg·m-3·d-1和50 d,日均沼气产量1 200 m3.
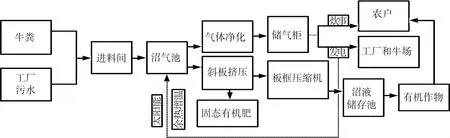
图1 海林农场沼气发酵系统生产模式
1.2 样品采集
样品采集于沼气池产气的稳定运行期,使用50 mL离心管于沼气池出料口处收集,厌氧封存后转移至冰盒,迅速带回实验室-20℃保存.
1.3 实验方法
1.3.1 微生物群落结构解析
样品自然融化后,以500 r/min离心30 s,弃沉淀后待用.基因组DNA应用Fast DNA SPIN Kit(Qbiogene Inc.)按照操作说明提取.
1.3.2 细菌群落结构解析
细菌16S rRNA V3高变区DNA扩增采用通用引物对341f和534r.引物序列如表1所示,下划线序列为454焦磷酸测序时区分各样品所加7个碱基的唯一编码序列标记.PCR扩增体系包括:模板DNA 50 ng,1X buffer,3 mmol/L MgCl2,每种引物10 pmol,0.4 mmol/L dNTP及1.25 U TaKaRa Ex Taq®HS.PCR扩增条件为94℃预变性5 min,95℃变性45 s,57℃退火30 s,72℃延伸45 s,30个循环,最后72℃延伸5 min.扩增产物使用FLX 454 System测序,由国家人类基因组南方研究中心协助完成.
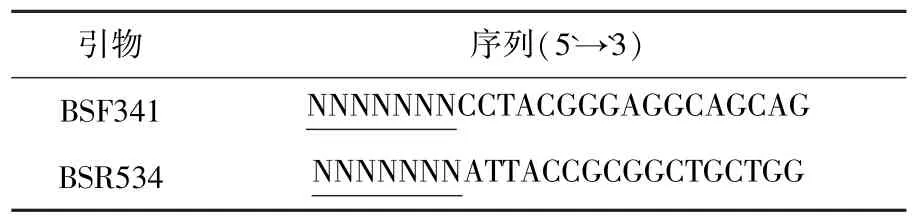
表1 16S rRNA细菌通用引物
所测得序列根据样品唯一的tag标记区分,使用RDP-II Classifier数据库将序列进行遗传分类(从Genus到Phylum)[8].相似矩阵(distance matrices)、聚类分析(cluster)、稀释曲线(rarefaction curve)、Shannon-Weaver指数、覆盖率(coverage estimator)、丰富度指数Chao1和ACE等应用mothur software计算(http://www.mothur. org/wiki/Sogin-data-analysis)[9].
1.3.3 产甲烷古菌群落结构解析
产甲烷古菌V2-V3区扩增使用通用引物对A109(T)-F和515GC-R[10].PCR扩增体系(50 μL)包括:模板DNA 100 ng,1X buffer,3 mmol/L MgCl2,20 pmol每种引物,0.4 mmol/L dNTP,1.25 U Taq DNA聚合酶.PCR扩增条件为94℃预变性3 min,94℃变性30 s,56℃退火30 s,72℃延伸45 s,30个循环,最后72℃延伸5 min.所有PCR反应使用GeneAmp PCR System 9700完成(Applied Biosystems,CA,USA).
DGGE应用30%~50%的梯度胶,凝胶PCR产物点样后于60℃、75 V电泳16 h后银染.将切胶后的条带样品回收、纯化,PCR产物与pGEM-T载体连接,转化JM109,随机挑取克隆,进行转化子鉴定后测序.所得序列通过Genbank数据库的BLASTX功能进行分析比对.序列的线性及多重比对使用Clustal-X软件完成,系统发育进化树使用Mega 4.0以Neighbor-Joining算法构建,自评1 000次[11].
2 结果与讨论
2.1 沼气池细菌群落多样性分析
为获取更多沼气池稳定运行期细菌群落多样性信息,利用454高通量测序技术对细菌16S rRNA的V3高变区进行扩增,序列比对后分析种群多样性和结构组成.经序列质量筛选,去除碱基错配、缺失、含(N)序列及短序列等,获得高质量序列1 297条.
2.1.1 细菌多样性评价
通量测序的数据分析一般将不同序列在同一相似水平下归类于同一操作分类单元(operational taxonomic unite,OTU).如表2所示,当序列相似度为95%和97%时,分别产生581个和666个OTU,覆盖率分别为66.8%和60.4%,说明沼气池稳定运行时至少存在581个细菌属和666个细菌种,细菌群落多样性非常丰富.丰富度指数Chao1和ACE是反映物种群落丰欠状况的指标,本研究计算结果显示,在属分类水平沼液样品Chao1和ACE分别为1 937和3 745.丰富度指数分析同样表明,测序数量未能达到饱和,继续扩大测序通量仍可发现新的物种.
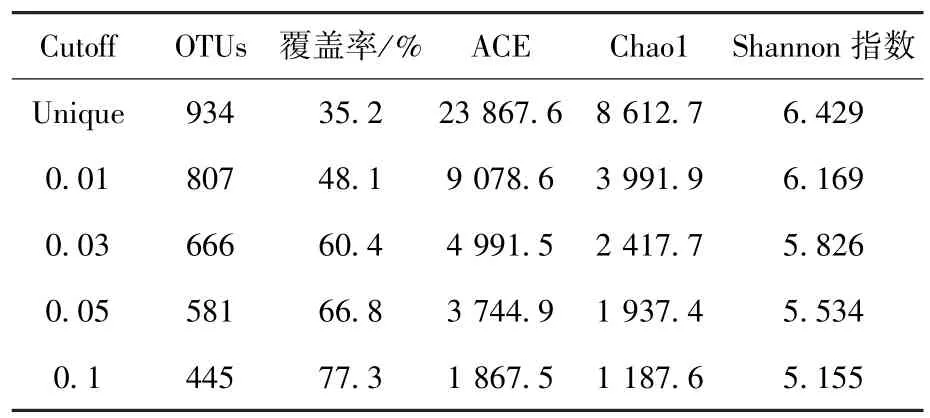
表2 细菌在不同分类水平的多样性及丰富度指数统计
图2为沼液样品细菌群落在不同分类水平下的稀释曲线,取95%为置信区间,以每次随机挑选10条序列进行累加的方法计算.可以看出,起初随着测序数量的增加,OTU数量迅速增加;随着测序序列数量的进一步增大,各分类水平下OTU数目的增加趋于平缓,但仍未达饱和.稀释曲线的平滑程度越高,表明对环境样品微生物的覆盖率越高.

图2 沼液样品稀释曲线
2.1.2 细菌群落组成的系统发育分析
本研究从沼气池沼液样品中获得1 297条高质量有效微生物序列,根据系统发育分析,序列相似度cutoff=0.3时的聚类分析,可产生666个OTU,即沼气池中存在666个细菌种(species),相比传统常用微生物研究的分子生物学手段,DGGE和克隆文库技术体现出巨大优势.由于传统分子生物学技术基于Sanger测序法,一个宏基因组样品聚类分析后获得的OTU数小于50,极大阻碍了准确了解群落结构的组成.本研究在种的分类水平上,获得的OTU数相比传统技术增加了10倍以上,充分显示出高通量测序技术的优越性.
如图3(a)所示,沼气池沼液样品细菌多样性十分丰富,在门分类水平上属于13个类群,主要包括:脱铁杆菌门(Deferribacteres)、揉膜菌门(Tenericutes)、绿弯菌门(Chloroflexi)、蓝藻门(Cyanobacteria)、Synergistetes门、放线菌门(Actinobacteria)、纤维杆菌门(Fibrobacteres)、疣微菌门(Verrucomicrobia)、绿菌门(Chlorobi)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、螺旋体门(Spirochaetes)和变形菌门(Proteobacteria).优势类群为Firmicutes、Bacteroidetes和Proteobacteria,相对丰度分别占细菌群落的46.39%、21.41%和18.98%.处于次优势地位的类群为Spirochaetes和Fibrobacteres,相对丰度分别占细菌群落的8.27%和3.17%.
如图3(b)所示,沼液样品在纲分类水平属于20个微生物类群,其中优势类群主要有(相对丰度>2.0%):梭菌纲(Clostridia)、拟杆菌纲(Bacteroidia)、ε-变形菌纲(Epsilonproteobacteria)、芽孢杆菌纲(Bacilli)、螺旋体纲(Spirochaetes)、β-变形菌纲(Betaproteobacteria)、纤维杆菌纲(Fibrobacteria)和γ-变形菌纲(Gammaproteobacteria),相对丰度分别占细菌群落的36.60%、19.11%、11.52%、8.79%、8.70%、5.29%、3.50%和2.05%.
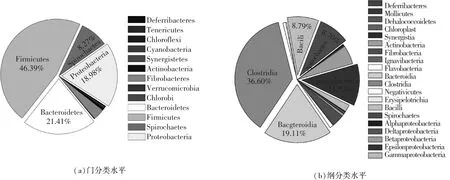
图3 沼气池细菌在门和纲分类水平的微生物组成
在属分类水平,优势类群主要包括(相对丰度>2.0%):Proteiniphilumsp.(7.33%)、Spirochaeta sp.(6.78%)、Wolinella sp.(5.86%)、Coprococcussp.(4.16%)、Arcobacetrsp.(3.86%)、Fibrobactersp.(3.47%)、Tetrathiobacter sp.(2.93%)、Lysinibacillus sp.(2.47%)、Bacillus sp.(2.24%)和Bacteroides sp.(2.16%)(表3).

表3 沼气池样品中相对丰度高于0.5%微生物OTU组成
2.2 沼气池产甲烷古菌多样性分析
为研究沼气池稳定运行期产甲烷古菌群落结构,提取沼液细菌基因组DNA,对产甲烷古菌16S rRNA V2-V3区PCR扩增后,扩增产物进行变性梯度凝胶电泳.DGGE图谱如图4所示,图谱中包含12个优势条带,对优势条带进行DNA胶回收后,进行基因克隆测序.由条带的相对丰度可知,Band 2、Band 3和Band 12是最优势类群.系统进化分析表明,沼气池产甲烷古菌主要分布在3个目,即甲烷八叠球菌目(Methanosarcinales)、甲烷微菌目(Methanomirobiales)和甲烷杆菌目(Methanobacteriales).核酸序列比对结果显示,沼气池产甲烷古菌包括Methanocorpusculum sp.、Methanosaetasp.、Methanobacteriumsp.及Methanosarcina sp.(表4).其中,Methanosaeta sp.和Methanosarcina sp.是沼气池内最优势产甲烷菌.

图4 沼气池产甲烷古菌V2-V3区PCR产物DGGE图谱
3 讨 论
深入解析生物反应器的微生物群落结构,可揭示其在运行过程中的运行特征及已知的功能微生物群落结构和未知群落之间存在的关系.应用454焦磷酸高通量测序技术研究沼气池稳定运行期沼液样品细菌群落结构,得到了较高的覆盖率,共获得1 297条高质量16S rRNA基因序列,覆盖率在属水平可达66.8%.相比传统分子生物学手段获得了更全面、准确的微生物群落信息.其中最优势类群属于Firmicutes(46.39%)、Bacteroidetes(21.41%)和Proteobacteria(18.98%),占序列总数的86%以上.在属的水平上,优势类群包括Proteiniphilumsp.(7.33%)、Spirochaetasp.(6.78%)和Wolinella sp.(5.86%),均属动物消化系统内常见微生物类群.由此可知,沼气池有机底物水解、产酸过程的主要功能微生物来源于牛消化系统内的细菌类群,并未演替为具有较强有机物降解能力的Bacillus sp.、Clostridium sp.和Acinetobacter sp.等,以及挥发酸转化能力较强的Syntrophomonassp.、Ruminococcussp.和Desulfotomaculum sp.等微生物类群[12-14].由于沼气池采用的是单相混合发酵工艺运行,挥发性脂肪酸的转化速率仍是产甲烷效率的限速步骤.此外,以Proteiniphilum sp.、Spirochaeta sp.和Wolinella sp.为优势类群的群落结构,影响了底物的降解和产酸速率,这也是造成沼气池产气量不高的主要原因之一.
很多研究证实,Bacteroidetes包括很多具有降解长链脂肪酸功能的微生物类群,在降解长链脂肪酸时具有重要作用[15].一些长链脂肪酸,如十二烷酸、油酸、辛酸和豆蔻酸等,尤其是十二烷酸是毒害作用最强的长链脂肪酸.Wang等[16-17]在CSTR厌氧消化牛粪、牛粪与少量秸秆混合产甲烷过程中以及处理一些富含糖类物质的废弃物时发现Bacteroidetes为优势菌群.当OLR增加时Clostridiaceae一类菌群增加,这表明系统内微生物代谢由脂肪酸转化转变为有机底物的降解.Lee等[18]在应用焦磷酸通量测序技术对大规模混合及两相工艺中温处理剩余污泥产甲烷研究时发现,优势种群为Proteobacteria(20.5%)、Bacteroidetes(19.7%)、Firmicutes(17.8%)和Chloroflexi(4.8%).Godon等[19]研究应用蒸酒废物产甲烷时发现,最优势类群属Firmicutes,其次为Bacteroidetes和Proteobacteria.这一结论与本研究相似,Bacteroidetes在很多中温发酵产甲烷系统中均为优势种群,表明与有机物降解的水解、酸化过程紧密相关[20].
应用PCR-DGGE对沼气池产甲烷古菌多样性进行分析,结果表明,优势产甲烷类群主要分布在Methanosarcinales、Methanomirobiales和Methanobacteriales.由条带相对丰度可知,Methanosaeta sp.和Methanosarcina sp.是沼气池内优势产甲烷古菌,均为乙酸营养型产甲烷古菌.由此可知,沼气池以利用乙酸产甲烷途径为主要代谢方式.利用H2及甲酸盐为电子供体的Methanobacterium sp.和Methanocorpusculum sp.,在维持乙酸营养型产甲烷系统稳定运行中也具有重要作用.Rastogi等[21]研究夏季以牛粪为发酵底物产沼气系统中产甲烷菌群多样性时发现,优势产甲烷古菌为Methanomicrobiales、Methanosarcinales和Methanobacteriales,分别占序列总数的41.7%,30%和19%.此外,Methanosarcina sp.稳定存在于厌氧发酵系统,也十分有利于低温条件下厌氧发酵系统的稳定运行.
由此可见,如何调控微生物成为降解和产酸能力较强的群落组成结构及活性较强的产甲烷菌类群,是提高系统运行效率的关键.为使厌氧消化系统高效运行及调控,可进行生物相的分离,使有机底物的降解和产酸过程与产甲烷过程分离,提高系统的稳定性.此外,通过调整运行参数、底物性质及投加外源菌群调控等策略,使产酸系统微生物类群由动物消化系统内微生物类群主导的组成过渡为对纤维类底物降解能力更强的群落组成,提高底物降解及挥发性有机酸的转化效率,同时适当调整产甲烷相的运行参数,提高产甲烷效率.
4 结 论
1)本研究利用454焦磷酸高通量测序技术,对北方规模最大的海林农场沼气池细菌群落进行多样性与结构解析,得1 297条高质量细菌序列,在属分类水平上的覆盖率为66.8%,至少存在581个细菌属.
2)聚类分析结果表明,沼气池细菌包括13个细菌门,最优势类群为Firmicutes,其次为Bacteroidetes和Proteobacteria.在属分类水平上,Proteiniphilum sp.为最优势微生物类群,其次分别为Spirochaeta sp.和Wolinella sp..
3)DGGE条带分析表明,沼气池产甲烷菌包括Methanocorpusculum sp.、Methanosaeta sp.、Methanobacterium sp.及Methanosarcina sp.,其中Methanosaeta sp.和Methanosarcina sp.是沼气池内优势产甲烷菌.
[1]RESCH C,BRAUN R,KIRCHMAYR R.The influence of energy crop substrates on the mass-flow analysis and the residual methane potential at a rural anaerobic digestion plant[J].Water Science and Technology,2008,57(1):73-81.
[2]DEMIRBAS M F.Current technologies for biomass conversion into chemicals and fuels[J].Journal of Energy Sources,2006,A28(13):1181-1188.
[3]任南琪,王爱杰,马放.产酸发酵微生物生理生态学[M].北京:科学出版社,2005.
[4]LO Y C,SARATALE G D,CHEN W M,et al.Isolation of cellulosehydrolytic bacteria and applications of the cellulolyticenzymesforcellulosicbiohydrogen production[J].Enzyme Microbial Technology,2009,44(6/7):417-425.
[5]任南琪,刘敏,王爱杰,等.两相厌氧系统中产甲烷相有机酸转化规律[J].环境科学,2003,24(4):89-93.
[6]NYANGA L K,NOTU M J R,GADAGA T H,et al. Yeasts and lactic acid bacteria microbiota from masau(Ziziphus mauritiana)fruits and their fermented fruit pulp in zimbabwe[J].International Journal of Food Microbiology,2007,120(1/2):159-166.
[7]赵光,马放,魏利,等.北方低温沼气发酵技术研究及展望[J].哈尔滨工业大学学报,2011,43(6):29-33.
[8]COLE J R,WANG Q,CARDENAS E,et al.The ribosomal database project:improved alignments and new toolsforrRNAanalysis[J].NucleicAcids Research,2009,37(1):141-145.
[9]SCHLOSS P D,WESTCOTT S L,RYABIN T,et al. Introducingmothur:open-source,platformindependent,community-supportedsoftwarefor describing and comparing microbial communities[J]. Applied and Environment Microbiology,2009,75(23): 7537-7541.
[10]LARKIN M A,BLACKSHIELDS G,BROWN N P,et al.ClustalWandclustalXversion2.0[J]. Bioinformatics,2007,23(21):2947-2948.
[11]KOICHIRO T,JOEL D,MASATOSHI N,et al. MEGA4:molecularevolutionarygeneticsanalysis(MEGA)software version 4.0[J].Molecular Biology Evolution,2007,24(8):1596-1599.
[12]LI T,MAZ'EAS L,SGHIR A,et al.Insights into networks of functional microbes catalysing methanization ofcelluloseundermesophilicconditions[J]. Environmental Microbiology,2009,11(4):889-904.
[13]WU C,LIU X,DONG X.Syntrophomonas erecta subsp.Sporosyntropha subsp.nov.,a spore-forming bacterium that degrades short chain fatty acids in coculture with methanogens[J].Systematic and Applied Microbiology,2006,29(6):457-462.
[14]MERLINO G,RIZZI A,VILLA F,et al.Shifts of microbial community structure during anaerobic digestion of agro-industrial energetic crops and food industry byproducts[J].JournalofChemicalTechnologyBiotechnology,2012,87(9):1302-1311.
[15]SHIGEMATSU T,TANG Y,KAWAGUCHI H,et al. Effect of dilution rate on structure of a mesophilic acetate-degradingmethanogeniccommunityduring continuous cultivation[J].Journal of Bioscience and Bioengineering,2003,96(6):547-558.
[16]HERNON F,FORBES C,COLLERAN E.Identification of mesophilic and thermophilic fermentative species in anaerobic granular sludge[J].Water Science and Technology,2006,54(2):19-24.
[17]WANG H,KATARIINA K.Microbial community structure in anaerobic co-digestion of grass silage and cow manure in a laboratory continuously stirred tank reactor[J].Biodegradation,2010,21(1):135-146.
[18]LEE S H,KANG H J.Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters[J].JournalofEnvironmental Monitoring,2012,14:1893-1905.
[19]GODON J J,ZUMSTEIN E,DABERT P,et al. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis[J].Applied and Environment Microbiology,1997,63(7):2802-2813.
[20]CHOUARI R,PASLIER D L,DAEGELEN P,et al. Novelpredominantarchaealandbacterialgroups revealed by molecular analysis of an anaerobic sludge digester[J].Environmental Microbiology,2005,7(8): 1104-1115.
[21]RASTOGI G,RANADE D R,YEOLE T Y,et al. Investigationofmethanogenpopulationstructurein biogas reactor by molecular characterization of methylcoenzyme M reductase A(mcrA)genes[J].Bioresource Technology,2008,99(13):5317-5326.
(编辑 刘 彤)
Analysis of microbial community in a full-scale biogas digester of cold region using high-throughput sequencing technology
ZHAO Guang1,2,MA Fang1,2,SUN Ting3,LI Shuben4,YOU Kong4,ZHAO Zhen1,2
(1.State Key Lab of Urban Water Resource and Environment,Harbin Institute of Technology,150090 Harbin,China;2.School of Municipal and Environmental Engineering,Harbin Institute of Technology,150090 Harbin,China;3.School of Chemical and Environmental Engineering,Liaoning University of Technology,121001 Jinzhou,Liaoning,China;4.Hailin Farm,150000 Mudanjiang,Heilongjiang,China)
To realize the stable and efficient operation of biogas digester in cold region,a combined regulation technique of microbial community should be established.The microbial community in the largest full-scale digester of Hailin Farm was investigated using 454 pyrosequencing technology and PCR-DGGE.The massively parallel sequencing technology was used to measure bacterial diversity of biogas slurry during a stable operation.A total of 1297 sequences were obtained,and the dominant bacteria were Firmicutes,Bacteroidetes and Proteobacteria,which accounted for 46.39%,21.41%and 18.98%,respectively.At genus level(the relative abundances>5.0%),Proteiniphilum,Spirochaeta and Wolinella were the abundant taxa.The diversitiesofmethanogenwereanalyzedusingPCR-DGGE,andthedetectedarchaeawere Methanocorpusculum sp.,Methanosaeta sp.,Methanobacterium sp.and Methanosarcina sp..Notably,the methane produced by acetoclastic methanogens,and dominant fermentative bacteria during the hydrolysis and acidogenesis were detected form animal digestive system.
biogas;microbial community;Hailin Farm;cow manure;454 pyrosequencing technology
X705
A
0367-6234(2014)04-0036-07
2013-03-20.
国家科技支撑计划专题项目(2012BAD14B06-04);辽宁工业大学教师科研启动基金资助项目(X201310).
赵 光(1980—),男,博士研究生;
马 放(1963—),男,教授,博士生导师.
马放,mafang@hit.edu.cn;孙婷,suntinghit@126.com.
猜你喜欢
土壤学报(2022年3期)2022-08-26
大自然探索(2022年5期)2022-07-11
知识就是力量(2022年6期)2022-06-16
煤气与热力(2021年12期)2022-01-19
环境卫生工程(2020年6期)2020-12-30
中国资源综合利用(2018年12期)2019-01-08
浙江农业科学(2016年11期)2016-05-04
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
河北建筑工程学院学报(2015年2期)2015-04-29
河北建筑工程学院学报(2015年4期)2015-03-24
