
脑利钠肽对兔心肌梗死后缝隙连接蛋白重构及炎症因子的影响*
2014-06-13 01:17:10王伯松
山东第一医科大学(山东省医学科学院)学报 2014年12期
王伯松
(泰山医学院附属泰山医院心内科,山东 泰安 271000)
心肌缺血相关的室性心律失常是导致猝死的重要原因之一。缝隙连接介导心肌细胞间的电藕联,缝隙连接的正常表达与分布,对于维持心脏电活动与机械活动同步有重要作用。缝隙连接蛋白Cx43的减少及分布紊乱均会导致缺血区心肌电传导异常,诱发心律失常[1]。脑利钠肽(BNP)是由心室分泌的多肽类的心脏激素,心肌损伤后伴随炎性反应的发生, BNP对此过程可能发挥调控作用。rhBNP是人工合成产物,与BNP作用机制相同,具有利钠、扩血管、抑制交感神经系统及肾素-血管紧张素-醛固酮(RAAS)系统活性,增强心肌抗缺血、抗缺氧能力,也能降低心律失常的发生率[2]。以往研究发现脑利钠肽能减少炎性细胞因子释放,抑制心室重构[3],但是具体机制尚不清楚。本实验旨在通过构建兔心肌梗死模型,观察rhBNP对于心肌梗死后炎症反应及缝隙连接蛋白数量、分布的影响。
1 材料与方法
1.1动物模型的制备与分组
58只新西兰大白兔(普通级,2.0~2.5kg,雌雄不限),由山东中医药大学实验动物中心提供。随机分为假手术组(Sham组)18只,心肌梗死组(MI组)20只, 心肌梗死后人重组脑利钠肽干预组(rhBNP组)20只。
兔心肌梗死模型制备:3%戊巴比妥钠(40 mg/kg)麻醉,常规消毒后沿胸骨中线切开皮肤,暴露胸骨及肋软骨。贴紧胸骨左缘剪断第4~5肋骨,暴露纵隔及心脏,剪开心包膜,暴露出冠状动脉。于左心耳下缘至心尖部的中下1/3处用6/0线穿过心肌浅层缝扎前降支,记录心电图。心电图示ST段抬高,结扎水平以下心肌局部颜色变暗,搏动减弱,观察20 min后逐层关闭胸腔。假手术组,手术过程同上只穿线但不结扎冠状动脉。术后给予青霉素(80万单位/d)注射3天,自主饮食(水)。
MI模型制备后,48只兔存活,Sham组(n=16),MI组(n=16),rhBNP组(n=16)。rhBNP组自术后即刻给予rhBNP(0.01 μg/kg/min)持续静脉泵入24 h;MI组、Sham组给予同等剂量的生理盐水静脉泵入;所有兔同期喂养8周。
1.2电生理实验
1.2.1测定心肌有效不应期(ERP) 术后8周所有存活兔以3%戊巴比妥钠(35 mg/kg)麻醉,心电监护,开胸,缝制心包吊床。起搏电极,直径0.3 mm ,两针相距0.3 cm,在梗死灶周(梗死苍白边缘3 mm)刺入2 mm,Sham组起搏电极放置在心脏相应部位,应用心脏电生理刺激仪(DF-5A型,苏州市东方电子仪器厂)以高出窦性频率10%的频率起搏,脉宽0.1 ms, 电压5.0 v,确定起搏有效后,逐渐降低起搏电压,测定起搏阈值,以起搏阈值的两倍为输出电压,脉宽0.1 ms ,以S1S2 逆扫,每次递减5 ms,测定ERP。
1.2.2程序电刺激诱发室性心律失常
1.2.2.1刺激终点 ①诱发出持续性室性心动过速(VT)或心室颤动(Vf); ②完成所有操作仍未诱发出室性心律失常。
1.2.2.2刺激方案 ①行S1S2刺激,以5ms逐步递减直至诱发VT/Vf或心肌不应期;②以S1S2刺激如未诱发出室速或室颤,则在不应期的基础上增加30ms,加发S3重复递减刺激,直至诱发VT/Vf或心肌不应期;③S1S1持续刺激8 min。如未诱发出室性心律失常,停止刺激5分钟后重复上述步骤。
1.3免疫荧光分析
新鲜的标本冰冻切片后(取材部位均与上述电生理指标测定位置一致),10%中性甲醛固定制作蜡块,制成4 μm切片。采用SABC法进行免疫组织化学检测。阴性对照用PBS代替一抗。滴加10%羊血清室温孵育20 min,滴加1∶100稀释的Cx43多克隆抗体(美国Millipore公司), 4 ℃孵育过夜,PBS洗3 min×3次,滴加1∶500稀释的异硫氰酸荧光素标记的IgG抗体(IgG- FITC), 37 ℃孵育20 min, PBS洗3 min×3次,缓冲甘油封片。 以Zeiss LSM510激光共聚焦显微镜观察,以488 nm波长激发FITC,发生光为519 nm的绿光。每张切片取4~5个视野,选择细胞纵行走向区域逐层扫描获取图像。
1.4IL-1β、TNF-α mRNA的表达水平的检测
2017年,国内数字出版行业的市场营业收入总额达到7071.93亿元,国内互联网期刊出版行业的市场营业收入总额达到20.10亿元,较2016年上涨了14.68%。
按照Trizol试剂盒(Invitrogen公司)提供的方法提取梗死灶周心肌总RNA。根据Genbank查询目的基因序列,Primer5.0设计引物,GADPH为内参照,GAPDH引物序列;上游5’ GCGCCTGGTCACCAGGGCTGCTT 3’,下游5’ TGCCGAAGTGGTCGTGGATGCCT 3’,扩增目的片段465bp。1)目的基因IL-1β引物序列:上游5’ATGGCAACTG TTCCTGAACTCAACT 3’,下游5’CAGGACAGGTA TAGATTCTTTCCTTT 3’,扩增目的片段257bp;2)目的基因TNF-α引物序列:上游5’CATCTTCTCA AAATTCGAGTGACAA 3’,下游5’TGGGAGTAGAC AAGGTACAACCC 3’,扩增目的片段175bp;按试剂盒说明一步法逆转录为cDNA。PCR反应体系:cDNA 10 μl、5×PCR缓冲液 10 μl、Tag酶0.25 μl,目的基因及GAPDH上游、下游引物各0.5 μl,加入双蒸水至总体积50 μl,95 ℃预变性5 min,扩增36个循环(条件:95 ℃变性30 s、53 ℃ 退火60 s、68 ℃延伸240 s,4 ℃ 5 min)。取PCR产物进行琼脂糖凝胶电泳,应用UVIpro凝胶图像分析系统进行荧光半定量分析,以目的基因与内参照物基因条带吸光度比值作为目的基因的相对表达水平。
1.5统计学分析
2 结 果
2.1动物的基本情况
术后8周Sham组存活16只, MI组存活16只,rhBNP组存活16只。MI组及rhBNP组心电图可见病理性Q波,梗死区心肌呈苍白色;Sham组冠状动脉穿线附近心肌无变化。
2.2电生理实验
2.2.2室性心律失常诱发率 进行程序电刺激,Sham组有1例诱发出VT,2例诱发出Vf;MI组有3例诱发出VT,5例诱发出Vf。而rhBNP组有2例诱发出VT,3例诱发出Vf,各组间均有显著性差异。(见表 1)。
2.3炎症因子 mRNA水平
MI组梗死灶周IL-1β mRNA、TNF-α mRNA水平均较Sham组相应部位均明显增高(P<0.01),rhBNP组较MI组IL-1β mRNA 、TNF-α mRNA水平减低(P<0.05)。(见表2)。
2.4免疫荧光及激光共聚显微镜分析
Sham组Cx43主要分布于心肌细胞的端-端连接处;MI组Cx43表达量明显显著减少,且分布发生改变,Cx43端-端连接减少,而侧-侧连接相对增多;而rhBNP组较MI组CX43表达增多,分布紊乱程度部分改善,趋于接近Sham组。(见图1~3)。
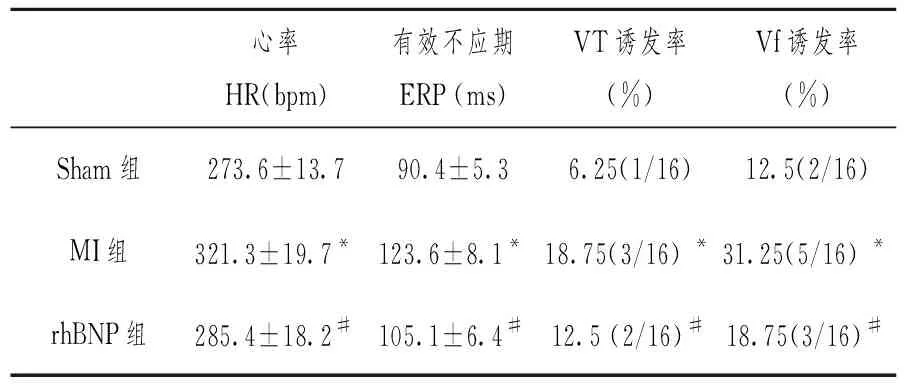
表 1 各组间电生理数据比较
注:与Sham组相比,*P<0.01;与MI组相比,#P<0.05

表 2 炎症因子 mRNA 水平(RT- PCR)
注:与Sham组相比,*p<0.01;与MI组相比,#p<0.05

图1 Sham 组Cx43分布均匀。

图2 MI组Cx43分布散乱。

图3 rhBNP组CX43分布紊乱程度得以改善。
3 讨 论
以往研究提示心肌细胞间电耦联的改变与心律失常的发生关系紧密,缝隙连接的数量及空间分布决定了细胞间在各方向上传导速度的不同,缝隙连接在空间上的重新分布造成了电传导各向异性的改变,而缝隙连接的数量改变或功能下降会使动作电位传导的速度减慢,增加电不稳定性增加导致心律失常易于发生。Perter[4]等通过对狗心肌梗死灶的心外膜下边缘区尚存活心肌进行研究,发现Cx43 的降解和分布的改变与心律失常的发生有着密切的关系,并且通过标测直接证实了Cx43分布改变与折返环路形成的关系,从分子水平揭示了心律失常发生的解剖学基础。
本研究观察梗死周边区Cx43减少,室颤阈值随之明显降低,提示心肌缺血破坏了正常的缝隙连接。可能有多种机制参与了这个过程,比如细胞内酸性物质堆积,氧自由基的大量生成等。神经内分泌因素和细胞因子激活的多个复杂信号传导通路介导了心肌缝隙连接重构。心肌梗死后梗死周边区炎症因子如IL-1β、TNF-α和ET-1表达显著增高[5]。TNF-α、IL-1可通过各自的信号传导通路影响Cx43数量和功能[6,7]。 并能够加剧心梗Cx43的降解[8]。 梗死灶周Cx43数目的下降会造成局部心肌电传导速度减慢,使缺血心肌与正常心肌之间电传导速度的差异增大,易于发生折返性心律失常。由此可以推测Cx43的减少可能是缺血诱发心律失常的一个重要因素。
rhBNP能够抑制RAAS和ET-1分泌,拮抗神经内分泌系统过度激活产生的心脏毒性[9],rh-BNP 可以使肾上腺的去甲肾上腺素分泌率明显下降,而不引起反射性交感神经兴奋及心率增加,还可直接抑制心脏交感神经活动[10]。持续而严重的心肌缺血在较短时间内造成心肌细胞的大量坏死,炎性细胞浸润并释放大量的炎症促进性的细胞因子。Chiurchi等[11]研究表明, BNP可调控THP-1人巨噬细胞炎性因子的产生,如活性氧、活性氮、白三烯B4受体、前列腺素E2以及TNF-α、IL-12和IL-10。炎症介质可能通过心肌细胞坏死、凋亡引起纤维化而参与心力衰竭的发生和发展。心肌梗死区、梗死周边区和非梗死区都存在着心肌细胞凋亡,心肌细胞凋亡也能促进Cx43的降解。以往研究发现脑利钠肽能显著抑制心肌纤维化[12]。
BNP 可能对心梗后心肌的急性炎症过程产生影响,进一步对心肌的炎症性损伤产生保护作用。rhBNP组的Cx43的免疫荧光检测显示BNP干预可降低梗死灶周炎症因子的表达,抑制心梗后Cx43的降解,促使Cx43的表达和分布趋与正常化,使得局部电传导的破坏减轻,正常心肌与缺血心肌之间缝隙连接的数目差距减小,电传导的一致性可能有所恢复,这有利于打破折返环,抑制心律失常的发生。从而推测BNP这种部分逆转缺血对缝隙连接的损伤作用,可能参与了BNP抑制心肌梗死后室性心律失常发生的过程。
[1] Peter NS,Green CR,Pool-Wilson PA,et al.Cardiac arrhythmogenesis and the gap junction [J]. Mol Cell Cardiol, 1995,27:37-44.
[2] Savio P,Souza D,Derek M,et al.B-type natriuretic peptide limits infarct size in rate isolated hearts via KATP channel opening[J]. Sm J Physiol Heart Circ Physiol,2003,284(6):1592.
[3] Yu XY,Liu CZ,Wang JH, et al. Brain natriuretic peptide and left ventricular remodeling after emergency percutaneous coronary intervention in acute myocardial infarction patients[J]. Zhong guoWei Zhong Bing Ji Jiu Yi Xue, 2008, 20(4): 204-206.
[4] Peters NS,Coromilas T,Severs NJ,et al. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia[J]. Circulation,1997,95: 988-996.
[5] Yuan M J, Huang C X, Tang Y H, et al. A novel peptide ghrelin inhibits neural remodeling after myocardial infarction in rats [J]. Eur J Pharmacol,2009, 618(1-3):52-57.
[6] Eugenin EA,Branes MC,Bermam JW,Saez JC.TNF-α plus INF-γ induce connexin 43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses[J].J Immumol,2003,170(3):1320-1328.
[7] Tonon R,D Andrea P.The functional expression of connexin 43 in articular chondrocytes is increased by interleukin I :evidence for a Ca2+-dependent mechanism[J]. Biorheology. 2002, 39(1-2): 153-160.
[8] Hao J, Suzuki K, Lu Y, et al. Inhibition of gap junction-mediated intercellular communication by TNF-αincultured human corneal fibroblasts[J]. IOVS, 2005,46(4): 1195-1200.
[9] Yancy CW,Saltzberg MT,Berkowita RL,et al.Safety and feasibility of using serial infusions of nesititide for heart failure in an outpatient setting.(from the FUSIONI Trial) [J].Am J Cardiol,2004,94(5):595-601.
[10] Brunner-La Rocca HP,Kaye DM,Woods RL,et al.Effects of intravenous brain natriuretic peptide on regional sympathetic activity in patients with chronic heart failure as compared with healthy control subjects[J].J Am Coll Cardiol,2001,37: 1221-122.
[11] Chiurchi?V, Izzi V, DcAquilio F, et al.Brain Natriuretic Peptide (BNP) regulates the production of inflammatory mediators in human THP -1 macrophages[J]. Regul Pept, 2008, 148 (1-3): 26-32.
[12] Ogawa Y,Tamura N,Chusho H,et al. Brain nartiuretic peptide appears to act locally as an antifibrotic factor in the heart[J]. Can J Physio Pharmacol,2001,79: 723.
猜你喜欢
小学科学(学生版)(2019年10期)2019-11-16 08:55:04
新乡医学院学报(2019年8期)2019-09-07 02:35:14
中国环境监察(2017年5期)2017-10-23 05:26:48
电测与仪表(2016年14期)2016-04-11 12:34:22
罕少疾病杂志(2016年4期)2016-03-11 16:34:36
中国卫生标准管理(2015年18期)2016-01-20 09:27:06
中国卫生标准管理(2015年16期)2015-01-26 20:17:45
西南军医(2015年4期)2015-01-23 01:18:58
卫生职业教育(2014年9期)2014-02-16 07:23:14
中国医药指南(2013年9期)2013-06-28 17:17:47
