
随机突变提高单胺氧化酶活性
2014-06-09 12:35:36陈雪君马元慧邵建华赖敦岳王志国陈振明
生物工程学报 2014年1期
陈雪君,马元慧,,邵建华,赖敦岳,王志国,陈振明
1 杭州师范大学生命与环境科学学院,浙江 杭州 310036
2 杭州师范大学生物催化研究室,浙江 杭州 311121
3 浙江台州清泉医药化工有限公司,浙江 台州 317300
4 杭州师范大学衰老研究所,浙江 杭州 311121
随机突变提高单胺氧化酶活性
陈雪君1,马元慧1,2,邵建华3,赖敦岳2,王志国4,陈振明2
1 杭州师范大学生命与环境科学学院,浙江 杭州 310036
2 杭州师范大学生物催化研究室,浙江 杭州 311121
3 浙江台州清泉医药化工有限公司,浙江 台州 317300
4 杭州师范大学衰老研究所,浙江 杭州 311121
陈雪君, 马元慧, 邵建华, 等. 随机突变提高单胺氧化酶活性. 生物工程学报, 2014, 30(1): 109−118.
Chen XJ, Ma YH, Shao JH, et al. Increasing activity of a monoamine oxidase by random mutation. Chin J Biotech, 2014, 30(1): 109−118.
前期获得了一个对底物美西律具有一定活性的单胺氧化酶突变体A-1 (F210V/L213C)。为进一步提高其酶活性,利用MegaWHOP PCR构建了库容约为104的随机突变库。筛选后获得了一个最优突变酶ep-1,比活力为A-1的189%。选择性测定结果表明,酶的对映体选择性有较大提高,E值由101提高到282;动力学常数测定揭示,酶催化效率有较大提高,kcat/Km由0.001 51 mmol/(L·s)提高到0.002 89 mmol/(L·s)。和A-1酶相比,在所测定的11种胺类底物中,ep-1对其他7种底物的比活力有较明显提高,对其他4种底物的比活力变化不大。序列分析表明,ep-1的突变为T162A。分子动力学模拟结果提示,该突变主要通过修正通道氨基酸的二级结构和扩大活性口袋来发挥作用。
单胺氧化酶,易错PCR,酶活力,分子动力学模拟
美西律 (图1) 是一种钠通道阻断剂,临床上常以消旋的形式用作抗心律失常药和镇痛剂。药理学研究表明[1-2],美西律的R-型对映体优先和心脏钠通道结合;与S构型相比,(R)-美西律与人骨骼肌纤维的结合作用更强。因此,有必要开展美西律的手性合成研究[3]。
美西律对映体的合成有过多篇报道,其中包括消旋中间体的拆分,N-乙酰衍生物的酶水解以及多步立体选择性合成[4-6]。最近,Koszelewski等[7]利用ω-转氨酶进行了消旋美西律的去消旋化研究。通过一锅二步反应,底物浓度为28 mmol/L时,反应48 h后消旋美西律可被完全转化为(S)-或(R)-美西律,产物ee值可超过99%,得率97%;但是其中一个比较明显的问题是反应中需要用到多种酶,包括 (S) -ω-转氨酶、(R)-ω-转氨酶、氨基酸氧化酶和脱氢酶。
利用单胺氧化酶(MAO, EC 1.4.3.4)进行拆分是一种很有潜力的获得手性胺的方法[8-9]。单胺氧化酶为黄素酶,可选择性地将消旋胺的一个对映体氧化为亚胺,亚胺与酶解离后在水中分解为酮与氨(图1)。如果单胺氧化酶的对映体选择性足够高,反应速度较慢的对映体在反应速度快的对映体反应完全后成为光学纯的手性胺。利用来自黑曲霉的MAO, Turner等[10]在反应体系中加入硼烷这种简单廉价的还原剂,使“废物”亚胺被还原为消旋的胺,这些胺再进行第二次酶催化氧化反应。两个反应循环进行,最终可得到100%的光学纯手性胺[9]。这一体系是“一锅”式反应,而且反应不需要添加通常氧化还原酶所需要的辅助因子。另外,Turner等的研究证实[11-12],MAO-N在酶改造后有可能大幅度提高酶活力,并具备广泛的底物谱,其中包括环状二级胺和三级胺等。
MAO-N-D5是Turner实验室经过多轮定向进化获得的含5个突变位点的黑曲霉MAO突变子[8],具高度应用潜力。但是我们的前期研究表明,MAO-N-D5对消旋美西律基本无活性;经过基于CASTing的半理性定向进化研究,我们获得了一个有一定活性的单胺氧化酶突变体A-1 (F210V/L213C)。本研究以此为起点,探讨通过随机突变进一步提高酶活性的可能性。
1 材料与方法
1.1 基因、菌种与质粒
MAO-N-D5由南京金斯瑞公司优化合成;大肠杆菌Escherichia coli DH5α,Escherichia coli BL21 (DE3)和JM109(DE3)均为本实验室保存菌种;表达质粒pET-28a(+)购自Novagen公司。
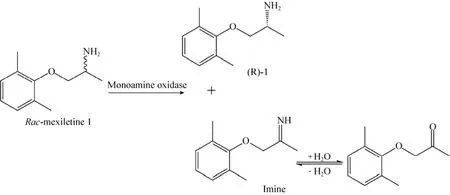
图1 美西律选择性拆分过程Fig. 1 Kinetic resolution process of rac-mexiletine to R-mexiletine by monoamine oxidase.
1.2 酶和试剂
限制性内切酶NdeⅠ、BamHⅠ购自英国NEB公司;DNA聚合酶KOD-plus-neo、高效连接酶ligation high和磷酸化酶PNK购自上海东洋纺生物科技有限公司;PCR cleaning试剂盒、质粒提取试剂盒、凝胶回收试剂盒和超级感受态细胞JM109 (DE3) 等购自杭州爱思进生物技术有限公司。
IPTG、卡那霉素和十二烷基磺酸钠购自上海生工生物工程有限公司;TBHBA、盐酸美西律、香草酸、DAB和4-AAP购自Sigma公司;辣根过氧化酶购自德国Roche公司;融合蛋白纯化试剂盒MagExtractor-His-tag购自美国GE公司;液相分析用试剂正己烷、三乙胺和乙醇等均为色谱纯试剂,购自百灵威科技有限公司。
1.3 易错PCR
以编码A-1的基因 (后均用a-1表示) 序列为模板,设计易错PCR引物。上游引物: 5'-ATGACGAGCCGCGACGGTTACCAAT-3';下游引物:5'-CAGACGGGCTTTCACTTCGCGTTT C-3'。易错PCR反应体系为:反应体积50 µL,MgCl27 mmol/L,MnCl2和dNTPs各0.2 mmol/L,dCTP和dTTP各1 mmol/L,10× PCR缓冲液5 µL,pET-28a-a-1 10 ng,上下游引物各0.3 µmol/L,Taq聚合酶1.25 U。易错PCR反应条件:预变性,94 ˚C 2 min;98 ˚C 20 s,58 ˚C 15 s,72 ˚C 2 min,28个循环;72 ˚C 5 min。
1.4 易错PCR突变文库的构建
反应体系组成:反应体积50 µL,纯化的易错PCR 产物0.5 µL, pET-28a-a-1 0.5 µL, MgCl21.5 mmol/L,dNTPs 0.2 mmol/L, 10×缓冲液5 µL,KOD-plus-neo 1 µL。MegaWHOP PCR反应条件为:68 ˚C 5 min;预变性,94˚C 2 min;95 ˚C 20 s,55 ˚C 20 s,68 ˚C 180 s,25个循环;68 ˚C 5 min。其中反应体系先在68 ˚C保温5 min的目的是利用KOD聚合酶3'-5'外切酶活性,切除epPCR在扩增产物的3'端引入的A碱基[15]。
往上述50 µL MegaWHOP PCR产物中加入2 µL甲基化酶DpnⅠ,37 ˚C放置1.5 h。该步目的是去除甲基化的母链模板。然后取DpnⅠ消化的PCR产物5 µL,转化入超级感受态细胞JM109 (DE3);37 ˚C培养箱过夜后,冲洗平板上的菌体,提取质粒。
1.5 突变文库的筛选
1.5.1 平板初筛
配制平板筛选显色液[13]:往20 mL磷酸缓冲液 (100 mmol/L, pH 7.4) 加入1%高纯度琼脂糖,微波加热至琼脂完全溶解,冷却至50 ˚C后加入DAB至终浓度 0.5 g/L,缓慢搅动至DAB完全溶解后加入辣根过氧化物酶 (终浓度2 U/mL) 与目标底物 (终浓度10 mmol/L),45 ℃温浴待用。
筛选过程[13]:将1.4中获得的随机突变质粒5 µL转化入BL21 (DE3) 感受态细胞中,涂板于5个覆盖有硝酸纤维素膜的含卡那霉素抗性的LB平板上,37 ˚C培养8–10 h;然后将长有菌落的硝酸纤维素膜揭下,覆盖于含1 mmol/L IPTG的LB平板上,20 ˚C培养14 h;揭下硝酸纤维素膜,平铺于灭菌的干净平板底部,将冷却至40 ˚C左右的平板显色液迅速倒入平板内,使显色液完全浸没硝酸纤维素膜;待显色缓冲液凝固后,将平板放入37 ˚C培养箱培养,每0.5 h观察一次;当出现棕色菌落时,用灭菌牙签挑取,接种,保存备用。
1.5.2 96孔板复筛
配制显色液[14]:100 mmol/L磷酸缓冲液(pH 7.4),4-AAP(4-aminoantipyrine) 7.5 mmol/L,0.2% (W/V)香草酸,辣根过氧化物酶4 U/mL,盐酸美西律10 mmol/L。
筛选过程[14]:将初筛得到的菌落接种于2 mL LB液体培养基中用IPTG诱导培养 (方法见1.6) 后收集菌体,洗涤后破碎取上清液;往透明96孔板加入显色液,每孔90 µL,再分别加入10 µL上清液;封口,37 ˚C培养箱中放置20 min,观察颜色变化。
1.6 酶基因的测序、表达及酶纯化测序由南京金斯瑞公司完成。
诱导表达:将经初筛和复筛获得的目标菌落接种于LB培养基中,37 ˚C振荡培养至OD600约为0.4−0.6;添加IPTG至终浓度1 mmol/L,25 ˚C,200 r/min,诱导16 h,超声波破碎后取上清液,利用融合蛋白纯化试剂盒MagExtractor-His-tag对重组蛋白进行纯化,最后按Bradford法测定其蛋白质的含量。
1.7 酶活力测定
根据Braun等[15]报道的方法,测定纯化的单胺氧化酶的活力。其中酶反应液的配置过程为:5 mL磷酸缓冲液(1 mol/L, pH 7.6),500 µL 2,4,6-三溴-3-羟基苯甲酸(2%,溶于DMSO),37.5 µL 4-AAP (1 mol/L),50 µL辣根过氧化物酶(终浓度2 U/mL),20 µL消旋盐酸美西律(终浓度10 mmol/L),补足水至50 mL。往1 mL 酶反应液中加入10 µL纯化的酶液,37 ˚C反应10 min后,测定510 nm吸光值。酶活单位定义为:37 ˚C下每分钟催化生成1 µmol 过氧化氢所需的酶量为一个酶活力单位(U)。kcat和Km值的测定采用Linewaver-Burk 双倒数作图法, 选用的底物浓度分别为1、2.5、5、10、20、40和80 mmol/L。
1.8 单胺氧化酶拆分反应
拆分反应和样品制备:反应体系包含100 µL纯化的酶液,消旋美西律10 mmol/L,50 mmol/L磷酸缓冲液 (pH 7.6),总体积2 mL。37 ℃、200 r/min振荡反应24 h;取200 µL反应液,加入氯化钠至饱和,加入2 mL异丙醇,振荡,从盐水中萃取底物和产物;取出上层有机相,加入无水氯化钙固体颗粒干燥并用0.45 µm滤膜过滤后,进行液相分析。HPLC分析条件:手性色谱柱Chiralcel AS-H (Daicel Chemical Industries, Japan,φ0.46×25),流动相为正己烷/乙醇 (94/6, V/V),流速1 mL/min,柱温35 ˚C,检测波长274 nm。
1.9 分子机制分析
1.9.1 分子对接
应用分子对接软件AutoDock[16],针对MAO-N-D5单胺氧化酶晶体结构[17-18](PDB ID:2VVM) 和底物盐酸美西律 (R型和S型) 进行了分子对接研究,以揭示二者之间的作用模式。对接应用Lamarckian genetic algorithm,蛋白酶分子采用Kollman电荷[19-20],相互作用的格点空间以FAD末端为中心,大小为60×60×60,其他参数采用软件默认,分别进行150次独立的对接运算。对接结果按照其构象之间的均方根偏差RMSD进行分组,以1.0 Å为标准。最后选取含有构象最多的分组中对接自由能最高的构象作为单胺氧化酶和盐酸美西律潜在的结合构象。
1.9.2 分子动力学
为研究残基突变对蛋白酶三维结构的影响,我们以MAO-N-D5单胺氧化酶的晶体结构为基础,应用Chimera软件[21]分别构建了突变体A-1和ep-1的三维结构,然后应用Gromacs软件[22]分别对A-1和ep-1体系进行分子动力学研究,以得到平衡状态下两突变体的稳定构象。首先将两个体系分别置于大小为78 Å × 78 Å × 78 Å周期性水盒子中 (水分子采用TIP3P模型),并在AMBER99SB力场下相继应用最陡下降法和共轭梯度法分别对体系进行3 000步的能量最小化运算;然后进行模拟退火运算,使体系在300 ps内由0 K逐步升温到300 K;随后对体系进行200 ps的限制性动力学计算,以进一步优化溶剂环境;最后采用NPT系综分别进行20 ns的动力学运算,使体系达到动力学平衡状态。
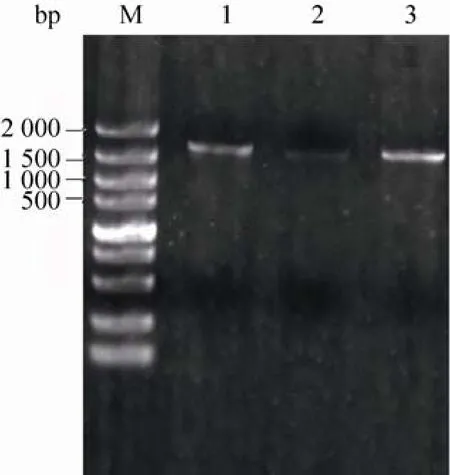
图2 易错PCR核酸电泳图Fig. 2 Result of epPCR. M: standard nucleic acid molecular weight; 1, 2, 3: three samples of parallel epPCR.
2 结果与分析
2.1 易错PCR突变库的构建
首先以A-1基因为模板,按照易错PCR常规条件 (高浓度Mg2+和不均衡dNTPs浓度) 进行随机突变。易错PCR结果如图2所示,目的条带在1 500 bp附近,与理论值1 488 bp接近。
传统上,随机突变文库的构建过程涉及双酶切、回收、连接和转化等多个步骤,效率低下。本研究根据MegaWHOP[23]的原理,以易错PCR产物为引物,以携带a-1基因的质粒为模板,经过相对简单的PCR扩增和DpnⅠ处理,即可用于转化超级感受态细胞JM109 (DE3)。这种方法节约时间,减少并简化实验步骤,提高突变效率。取MegaWHOP PCR产物转化入超级感受态细胞JM109 (DE3),培养过夜,收集菌体后提取质粒,即获得随机突变库。质粒检测结果如图3所示。
2.2 突变库的筛选
2.2.1 平板筛选
单胺氧化酶在催化胺的过程中产生微量过氧化氢,后者能被辣根过氧化物酶与二氨基联苯胺捕获,从而使有单胺氧化酶活性的菌落显棕色。根据这一原理筛选了大约10 000个菌落的随机突变库,挑取90个阳性菌落进行复筛。
2.2.2 96孔板筛选
单胺氧化酶催化反应所产生的过氧化氢在辣根过氧化物酶作用下,以4-AAP (4-aminoantipyrine)作为氢供体,被还原为水分子,4-AAP则以其氧化态存在,并与香草酸形成红色物质醌亚胺。通过红色的深浅,可初步判定单胺氧化酶活力的高低[24]。根据这一原理在96孔板上进行复筛,挑选颜色最深的10个突变菌株,进行下一步的工作。
2.3 突变酶的活力测定及酶催化性质的初步分析
将96孔板筛选获得的10个突变株,进行基因表达和蛋白纯化后测定比活力。选出比活力最高的6个菌株进行DNA测序,结果表明有3个突变株发生了突变,其中ep-1的突变为T162A, ep-7为W262L/D287N, ep-10为T324A。这3个突变株的相对比活力测定结果 (表1) 表明:ep-1的比活力比A-1提高了89%;另两个菌株则差别不大,甚至有所下降。
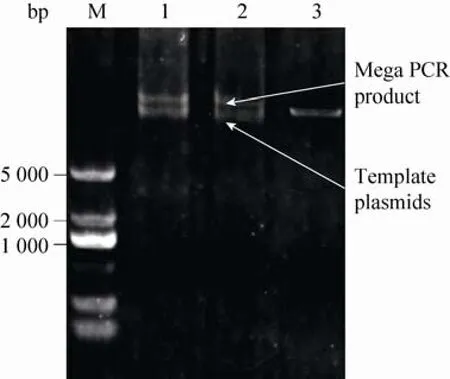
图3 Mega PCR核酸电泳图Fig. 3 Result of Mega PCR. M: nucleic acid marker; 1, 2:two samples of Mega PCR;3:plasmid pET28a-MAO-N-D5.
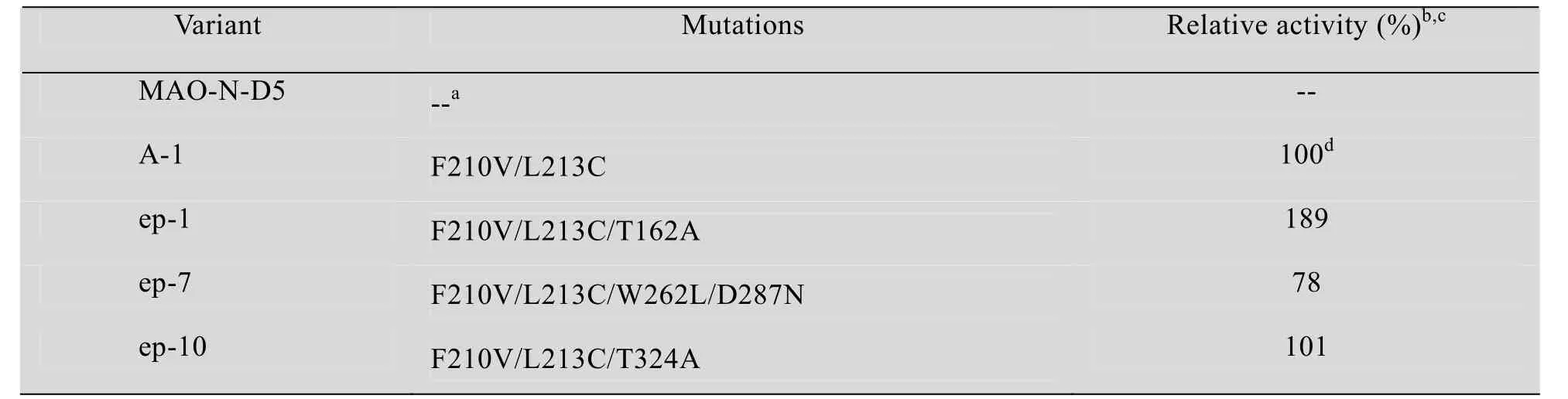
表1 突变菌株的活力测定Table 1 Specific activities of wild-type and mutant MAO enzymes
为验证筛选到的突变株的手性选择性,利用纯化酶进行消旋美西律的拆分反应。结果 (表2) 显示,ep-1是效果最好的突变子,对映体过量值和出发酶A-1一样,均在99%以上,表征对映体选择性的E值则由101提高到282;另外两个突变酶的选择性则较A-1有所下降。
以A-1为对照,进一步测定突变子ep-1的动力学参数。测定结果表明(表3),与A-1相比,ep-1的Km值由60.74 mmol/L降低到35.94 mmol/L;ep-1的kcat值有所增加,但是并不明显;kcat/Km由0.001 51 mmol/(L·s) 提高到0.002 89 mmol/(L·s)。
2.4 突变酶的底物特异性分析
以11种胺类物质为底物,包括非手性胺、手性一级胺、二级胺、芳香胺和脂肪胺,测定MAO-N-D5、A-1和ep-1三种酶的底物特异性,结果见表4。可以看出,ep-1的相对活力总体比A-1活力高。与未突变的MAO-N-D5相比,A-1与ep-1对其中1、2、4和6四种底物的活力有较大提升,其他则变化不明显。
2.5 MAO-底物结合模式及突变体三维结构分析

表2 美西律拆分反应Table 2 Specific conversions of MAO variants to mexiletine enantiomers

表3 突变酶的动力学常数Table 3 Kinetic comparison of two mutant MAO enzymesa
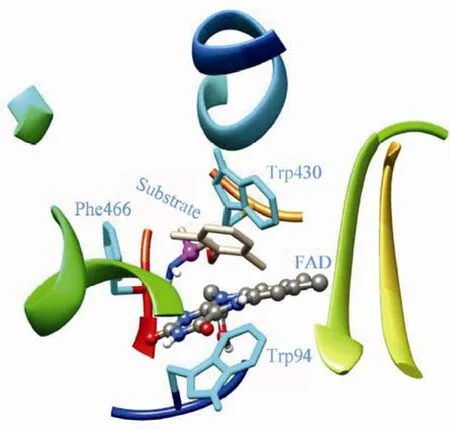
图4 MAO-N-D5与底物(S)-盐酸美西律的结合模式Fig. 4 Binding conformation of MAO-N-D5 and (S)-mexiletine.
应用分子对接研究得到了代表性底物盐酸美西律和MAO-N-D5的结合模式 (图4)。图中底物为盐酸美西律(S)-异构体,其手性C原子以粉红色圆球标出。据MAO对手性胺的催化机制可知,只有在这种取向下,底物手性C上的H原子才能有效地转移到辅酶FAD上,进而完成催化反应;R型异构体手性C上的H原子取向与S型相反,朝向远离FAD的方向,导致R型底物不能被有效催化,这也是MAO-N-D5蛋白酶对底物具有手性拆分能力的结构基础[17]。图4中还可以看到,以蓝青色标出的Trp430、Phe466和Trp94三个保守性残基将辅酶FAD和底物紧紧包围在一个较小的疏水性的口袋中,从而导致该蛋白酶对分子较大或极性较高的底物催化能力较差。
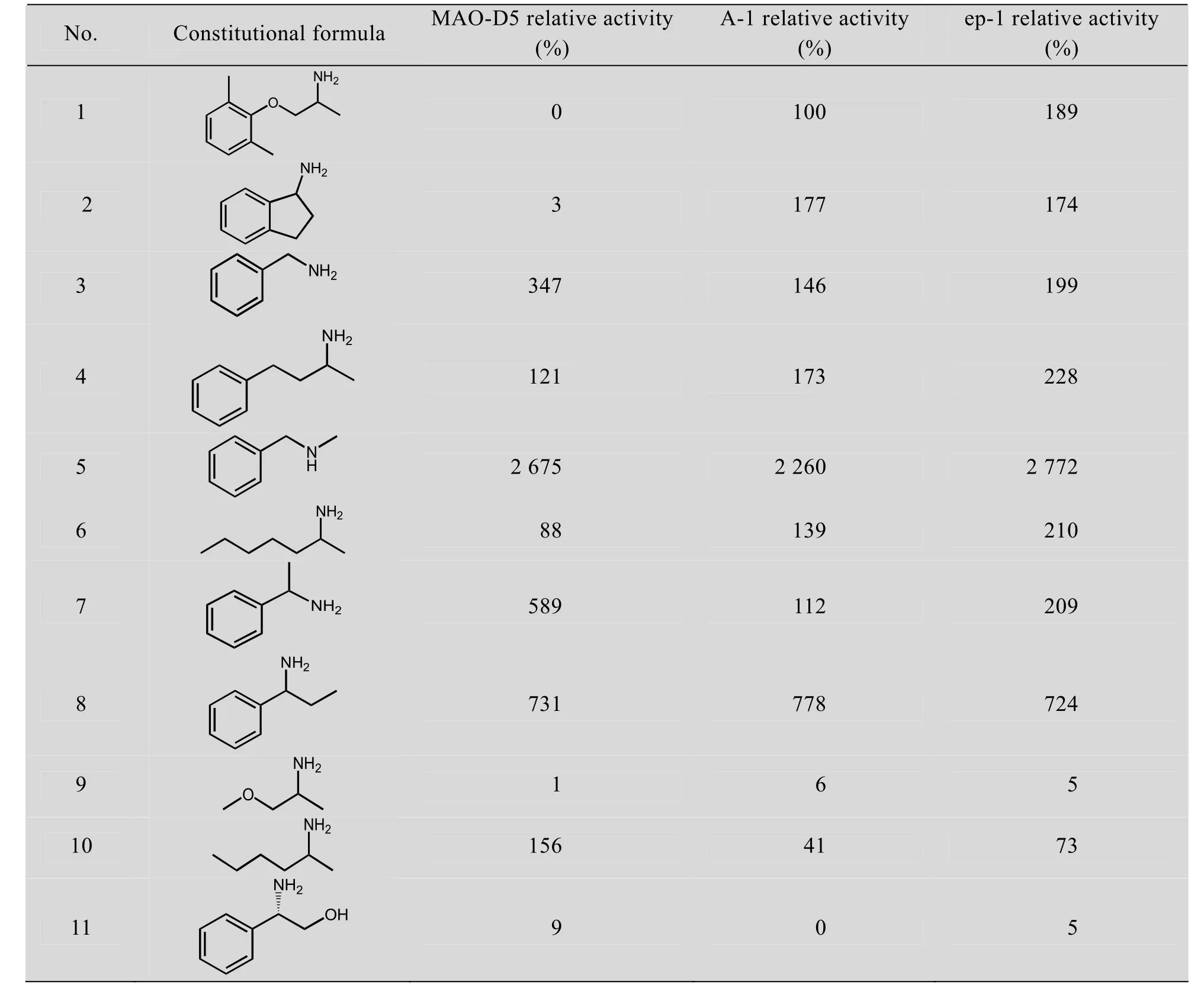
表4 突变酶底物特异性Table 4 Substrate specificity of MAO-N-D5 mutantsa,b
为了揭示残基突变如何在结构进而在功能上影响MAO-N-D5,我们将达到分子动力学平衡状态的突变体A-1和ep-1的三维结构与MAO-N-D5的晶体结构进行了叠加,其催化中心附近残基 (以辅酶FAD末端异咯嗪N5原子为中心,周围10 Å以内的所有残基) 的叠加情况见图5。由图可知,3个结构大部分结构均能较好的重叠 (MAO-N-D5、A-1和ep-1的结构分别标记为黄色、绿色和红色),表明相关突变没有导致MAO-N-D5的结构产生巨大变化。但是,图中有两个值得注意的地方:其一,蓝青色Trp430、Phe466和Trp94的位置。每个突变体上3个残基均比突变前更向外扩展,其中以ep-1在Trp94 loop上的表现最为明显,这表明两个突变体A-1和ep-1能够依次容纳更大的底物;其二,圆桶中紧邻催化中心的结构。该结构处在底物进出活性口袋的通道上,在MAO-N-D5为柔性较低的α-helix,在A-1中为loop和α-helix的结合,而在ep-1中则全部为柔性较高的loop结构。显然loop结构在酶结合较大的底物分子时较α-螺旋更为有利。正是以上两点结构上的变化为A-1和ep-1在催化盐酸美西律及类似底物时表现出更强的催化能力创造了结构基础。图中并未标出第210、213和162号突变残基,这是由于3个残基距离FAD末端异咯嗪部分较远,超出了催化中心的范围。这种结构特征表明残基突变并非与活性中心残基相关,而是通过改变其所属的二级结构来扩大活性口袋从而间接发挥作用的。
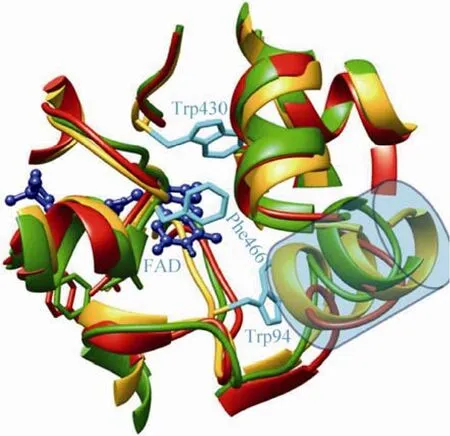
图5 突变体A-1 (绿色) 和ep-1 (红色) 与MAO-N-D5 (黄色) 催化中心三维结构的叠加Fig. 5 3D superimposition of catalytic centers of A-1, ep-1 and MAO-N-D5.
3 讨论
经过平板初筛、96孔板复筛及酶活测定,获得了1个突变子ep-1,其所包含的突变T162A使酶活提高了89%。选择性测定结果显示,ep-1的对映体过量值和A-1相似,ees均在99%以上;表征对映体选择性的E值则有较大幅度的提高。这说明T162A突变提高了酶催化反应的选择性。
动力学测定结果表明,ep-1的Km值明显降低, 表明ep-1对底物的亲和力有所提高,表征催化底物氧化的能力的kcat值则增加不明显。综合两个结果,T162A突变显著提高了酶的催化效率 (kcat/Km)。分子对接和分子动力学研究表明T162A突变在稳定保持活性中心残基构象的基础上,使得活性通道氨基酸的二级结构由A-1的loop-helix结构转变为完全的高柔性loop结构,从而为底物的高效结合与催化创造了条件。
尽管ep-1比A-1的比酶活提高了89%,但是实际上仍只是具备了初步活性,离工业化应用所需要的酶活还有较长的距离[25],需通过进一步的优化进行改造。
REFERENCES
[1] Turgeon J, Uprichard ACG, Bélanger PM, et al. Resolution and electrophysiological effects of mexiletine enantiomers. J Pharm Pharmacol, 1991, 43(9): 630–635.
[2] Luca AD, Natuzzi F, Falcone G, et al. Inhibition of frog skeletal muscle sodium channels by newly synthesized chiral derivatives of mexiletine and tocainide. N-S Arch Pharmacol, 1997, 356(6): 777–787.
[3] Franchini C, Carocci A, Catalano A, et al. Optically active mexiletine analogues as stereoselective blockers of voltage-gated Na+channels. J Med Chem, 2003, 46(24): 5238–5248.
[4] Carocci A, Catalano A, Bruno C, et al. Synthesis and in vitro sodium channel blocking activity evaluation of novel homochiral mexiletine analogs. Chirality, 2010, 22(3): 299–307.
[5] Höhne M, Bornscheuer UT. Biocatalytic routes to optically active amines. ChemCatChem, 2009, 1(1): 42–51.
[6] Koszelewski D, Clay D, Rozzell D, et al. Deracemisation of α-Chiral primary amines by a one-pot, two-step cascade reaction catalysed by ω-transaminases. European J Organic Chem, 2009, 2009(14): 2289–2292.
[7] Koszelewski D, Pressnitz D, Clay D, et al. Deracemization of mexiletine biocatalyzed by ω-transaminases. Org Lett, 2009, 11(21): 4810–4812.
[8] Turner N, Fotheringham I, Speight R. Novel biocatalyst technology for the preparation of chiral amines. Innov Pharma Technol, 2004, 4(14): 114–116.
[9] Carr R, Alexeeva M, Enright A, et al. Directed evolution of an amine oxidase possessing both broad substrate specificity and high enantioselectivity. Angew Chem Int Ed, 2003, 42(39): 4807–4810.
[10] Alexeeva M, Enright A, Dawson MJ, et al. Deracemization of α-methylbenzylamine using an enzyme obtained by in vitro evolution. Angew Chem Int Ed, 2002, 41(17): 3177–3180.
[11] Eve TSC, Wells A, Turner NJ. Enantioselective oxidation of O-methyl-N-hydroxylamines using monoamine oxidase N as catalyst. Chem Commun, 2007(15): 1530–1531.
[12] Dunsmore CJ, Carr R, Fleming T, et al. A chemo-enzymatic route to enantiomerically pure cyclic tertiary amines. J Am Chem Soc, 2006, 128(7): 2224–2225.
[13] Kim MJ, Kim WH, Han K, et al. Dynamic kinetic resolution of primary amines with a recyclable Pd nanocatalyst for racemization. Org Lett, 2007, 9(6): 1157–1159.
[14] Holt A, Palcic MM. A peroxidase-coupled continuous absorbance plate-reader assay for flavin monoamine oxidases, copper-cont aining amine oxidases and related enzymes. Nature Protocols, 2006, 1(5): 2498–2505
[15] Braun M, Kim JM, Schmid RD. Purification and some properties of an extracellular L-amino acid oxidase from Cellulomonas cellulans AM8 isolated form soil. Appl Microbiol Biotechnol, 1992, 37(5): 594–598.
[16] Morris GM, Goodsell DS, Halliday RS, et al. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Computational Chem, 1998, 19(14): 1639–1662.
[17] Atkin KE, Reiss R, Koehler V, et al. The structure of monoamine oxidase from Aspergillus niger provides a molecµLar context for improvements in activity obtained by directed evolution. J Mol Biol, 2008, 384(5): 1218–1231.
[18] Atkin KE, Reiss R, Turner NJ, et al. Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of variants of monoamine oxidase from Aspergillus niger. Acta Crystallogr Sect F: Struct Biol Cryst Commun, 2008, 64(3): 182–185.
[19] La Motta C, Sartini S, Mugnaini L, et al. Pyrido[1,2-a]pyrimidin-4-one Derivatives as a novel class of selective aldose reductase inhibitors exhibiting antioxidant activity. J Med Chem, 2007, 50(20): 4917–4927.
[20] Wang Z, Ling B, Zhang R, et al. Docking and molecular dynamics study on the inhibitory activity of coumarins on aldose reductase. J Phys Chem B, 2008, 112(32): 10033–10040.
[21] Pettersen EF, Goddard TD, Huang C, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem, 2004, 25(13): 1605–1612.
[22] Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible and free. J Comput Chem, 2005, 26 (16): 1701–1718.
[23] Kentaro M, Misa T. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques, 2002, 33(5): 1033–1038.
[24] Turner N, Fotheringham I, Speight R. Novel biocatalyst technology for the preparation of chiral amines. Innov Pharma Technol, 2004, 4(14): 114–116.
[25] Liang J, Lalonde J, Borup B, et al. Development of a biocatalytic process as an alternative to the (-)-DIP-Cl-Mediated. Org Process Res Dev, 2009, 14(1): 193–198.
(本文责编 陈宏宇)
Increasing activity of a monoamine oxidase by random mutation
Xuejun Chen1, Yuanhui Ma1,2, Jianhua Shao3, Dunyue Lai2, Zhiguo Wang4, and Zhenming Chen2
1 College of Life and Environmental Sciences, Hangzhou Normal University, Hangzhou 310036, Zhejiang, China
2 Laboratory of Biocatalysis, Hangzhou Normal University, Hangzhou 311121, Zhejiang, China
3 Zhejiang Taizhou Qingquan Medical & Chemical Co. Ltd., Taizhou 317300, Zhejiang, China
4 Institute of Aging Research, Hangzhou Normal University, Hangzhou 311121, Zhejiang, China
The monoamine oxidase mutant A-1 (F210V/L213C) from Aspergillus niger showed some catalytic activity onmexiletine. To futher improve its activity, the mutant was subjected to directed evolution with MegaWHOP PCR (Megaprimer PCR of Whole Plasmid) and selection employing a high-throughput agar plate-based colorimetric screen. This approach led to the identification of a mutant ep-1, which specific activity was 189% of that for A-1. The ep-1 also showed significantly improved enantioselectivity, with the E value increased from 101 to 282; its kinetic kcat/Kmvalue increased from 0.001 51 mmol/(L·s) to 0.002 89 mmol/(L·s), suggesting that catalytic efficiency of ep-1 had been improved. The mutant showed obviously higher specific activities on 7 of all tested 11 amines substrates, and the others were comparable. Sequence analysis revealed that there was a new mutation T162A on ep-1. The molecular dynamics simulation indicated that T162A may affect the secondary structure of the substrate channel and expand the binding pocket.
monoamine oxidase, error-prone PCR, activity, molecular dynamics simulation
July 25, 2013; Accepted: September 30, 2013
Zhenming Chen. Tel/Fax: +86-571-28869373; E-mail: zmchen05@gmail.com Zhiguo Wang. Tel: +86-571-28861733; E-mail: zhgwang@aliyun.com
Supported by: National Natural Science Foundation of China (No. 31100584).
国家自然科学基金 (No. 31100584) 资助。
猜你喜欢
高等学校化学学报(2024年2期)2024-03-06 06:31:12
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
中成药(2017年9期)2017-12-19 13:34:31
益寿宝典(2017年26期)2017-10-27 09:09:44
家庭医药(2017年7期)2017-07-15 23:30:57
中成药(2017年6期)2017-06-13 07:30:35
家庭医药(2017年13期)2017-03-25 04:36:34
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
