
噻吩结构与O2反应机理的理论研究
2014-06-07 05:55戴凤威邓存宝邓汉忠王雪峰
煤炭学报 2014年4期
戴凤威,邓存宝,邓汉忠,王雪峰,高 飞,张 勋
(1.辽宁工程技术大学安全科学与工程学院,辽宁阜新 123000;2.辽宁工程技术大学材料科学与工程学院,辽宁阜新 123000;3.辽宁工程技术大学矿业学院,辽宁阜新 123000)
噻吩结构与O2反应机理的理论研究
戴凤威1,邓存宝1,邓汉忠2,王雪峰1,高 飞1,张 勋3
(1.辽宁工程技术大学安全科学与工程学院,辽宁阜新 123000;2.辽宁工程技术大学材料科学与工程学院,辽宁阜新 123000;3.辽宁工程技术大学矿业学院,辽宁阜新 123000)
为研究煤自燃的反应机理,利用Gaussian 03程序,采用密度泛函理论(DFT)方法,在B3LYP/6-311G水平下研究煤结构中噻吩型有机硫与O2反应机理。由计算结果可知,反应物的初始连接方式有3种,其中Path 1的反应物经过过渡态TS1异构化为中间体IM1是无需克服势垒的放热过程,这说明煤中的噻吩结构无需从外界吸收热量,与氧气接触即可被氧化。此反应主要有6条反应路径,其中,Path 2是反应的主要反应路径,其速控步骤为过渡态TS6,需要克服的势垒为117.06 kJ/mol,其产物P2(C4H4O+SO)是反应的主要产物。
噻吩;反应机理;Gaussian 03程序;密度泛函理论
煤是一种具有芳香性的有机大分子,其有机质主要是由碳、氢、氧、氮和硫等元素组成。煤中的有机硫赋存形式主要有3种,分别为硫醇、硫醚和噻吩[1-2]。其中,噻吩型有机硫的含量非常多,在烟煤中的体现尤为突出[3-4]。随着科学技术的发展,科研工作者对煤炭自燃的研究越来越深入,将量子化学理论应用于煤炭研究已经成为了一种新的研究方法。谢克昌院士等采用量子化学原理对煤中含硫化合物的脱除进行了研究[5-6]。王继仁等采用量子化学原理,从不同角度研究了煤的活性基团对氧分子的物理吸附和化学吸附,研究结果表明:煤表面侧链基团对氧分子的吸附能大于苯环对氧分子的吸附能[7-9],煤发生氧化自燃主要是由于含N,S等活性基团被氧化引起。且噻吩的硫原子的两对孤电子中的一对与噻吩的两个双键共轭,从而形成离域π键,因此噻吩具有芳香性,其芳香性仅略弱于苯。噻吩的C—S键在光催化作用下易发生断裂[10-11],易于发生氧化或加氢反应。因此,煤结构中含硫结构与O2的反应机理已成为亟待深入研究的问题。本文采用Gaussian 03程序针对煤中噻吩型有机硫与氧气的反应机理进行了研究,为探索预防煤炭自燃技术奠定理论基础。
1 计算方法
全部计算利用Gaussian 03程序进行[12],采用密度泛函理论(density functional theory,DFT),首先在B3LYP/6-311G水平下对反应物、产物、中间体和过渡态分子进行几何优化,对每一个驻点进行振动频率分析,其中稳定构型的全部频率为正,而过渡态构型有且只有一个虚频[13-14]。然后,在同一水平下进行内禀反应坐标(intrinsic reaction coordinate,IRC)计算,确认反应物、中间体、过渡态和产物的相关性,证明过渡态的正确连接[15-16]。在以下的讨论中,将反应物(C4H4S+O2)的总能量设为能量零点。
2 结果与讨论
对于噻吩结构与O2的反应,计算所得的各条反应路径如下(Path 1~Path 6)所示。
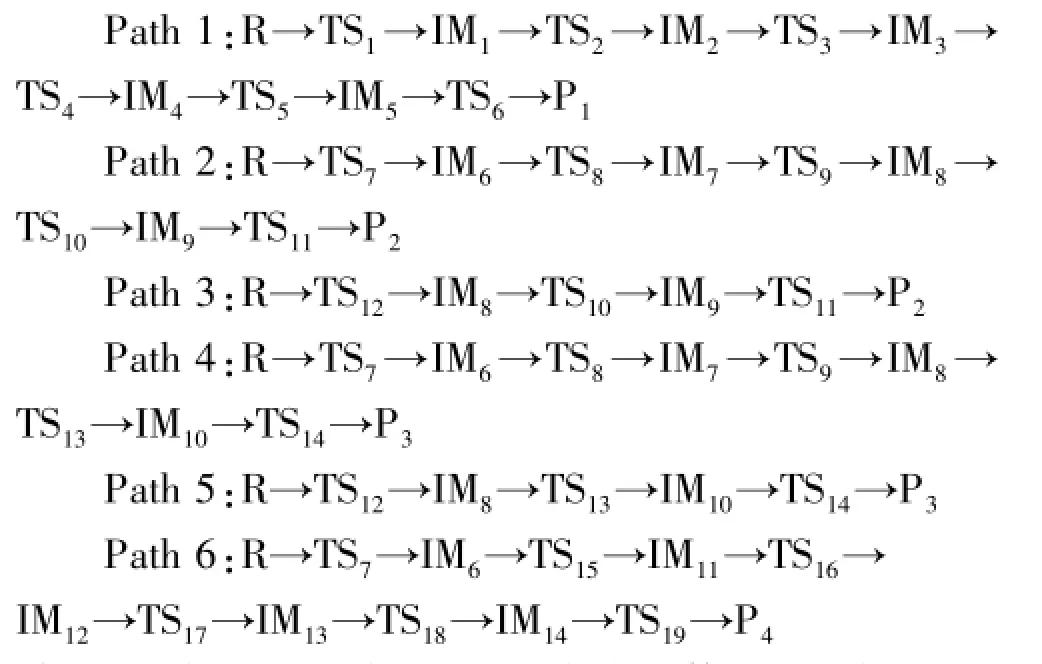
其中,反应物用R来表示,反应中间体用IM来表示,过渡态用TS来表示,产物则用P来表示。图1和图2分别列出了反应物、产物、中间体和过渡态的几何构型和参数。表1列出了反应物、产物、中间体和过渡态的总能量(total energies,TE)、零点振动能(zeropoint vibration energies,ZPVE)及相对能量(relative energies,RE)。其中,相对能量RE=相对反应物的总能量-相对反应物的零点振动能。根据各驻点的相对能量绘制噻吩结构与O2的反应势能面剖面图,如图3所示。从图3可看出,4种产物中P1,P2和P4的能量均比反应物低,热力学是可行的;而P3的能量比反应物高,热力学上是不利的产物。
2.1 反应物的初始连接
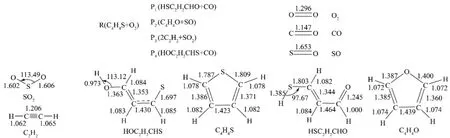
图1 在B3LYP/6-311G水平下计算反应物和产物的几何构型(键长单位:10-10m,键角单位:(°))Fig.1 Geometries of reactants and products at the B3LYP/6-311G level(Bond lengths:10-10meters,bond angles:degrees)
噻吩结构和O2反应的历程是复杂的多步反应,经历最初的结合以及随后的异构和离解,最终生成多种产物。根据Gaussian 03程序计算结果可知,噻吩结构与O2的结合方式主要有3种。第1种:两个O原子进攻C4H4S中S原子连接的两个C原子,形成不稳定的过渡态TS1,这一过程无需克服化学势垒,反而为进一步的异构化和分解反应提供了21.17 kJ/mol(图3)的能量,因此预测此种结合是此反应的主要反应入口;第2种:两个O原子的电子分别向C4H4S上S—C4键上S原子和C4原子靠近,氧分子的双键打开变成单键,与S原子和C4原子形成环状结构,经过过渡态TS7形成稳定的中间体IM6,此过程仅需克服15.40 kJ/mol的势垒,IM6的能量比反应物低149.70 kJ/mol,因此,这种结合方式也是很容易形成的;第3种:两个O原子共同进攻C4H4S的S原子,形成不稳定的过渡态TS12,经过过渡态TS12形成稳定的中间体IM8,这个过程需要克服的势垒为315.99 kJ/mol,很显然这种结合方式在反应路径的竞争中处于劣势。
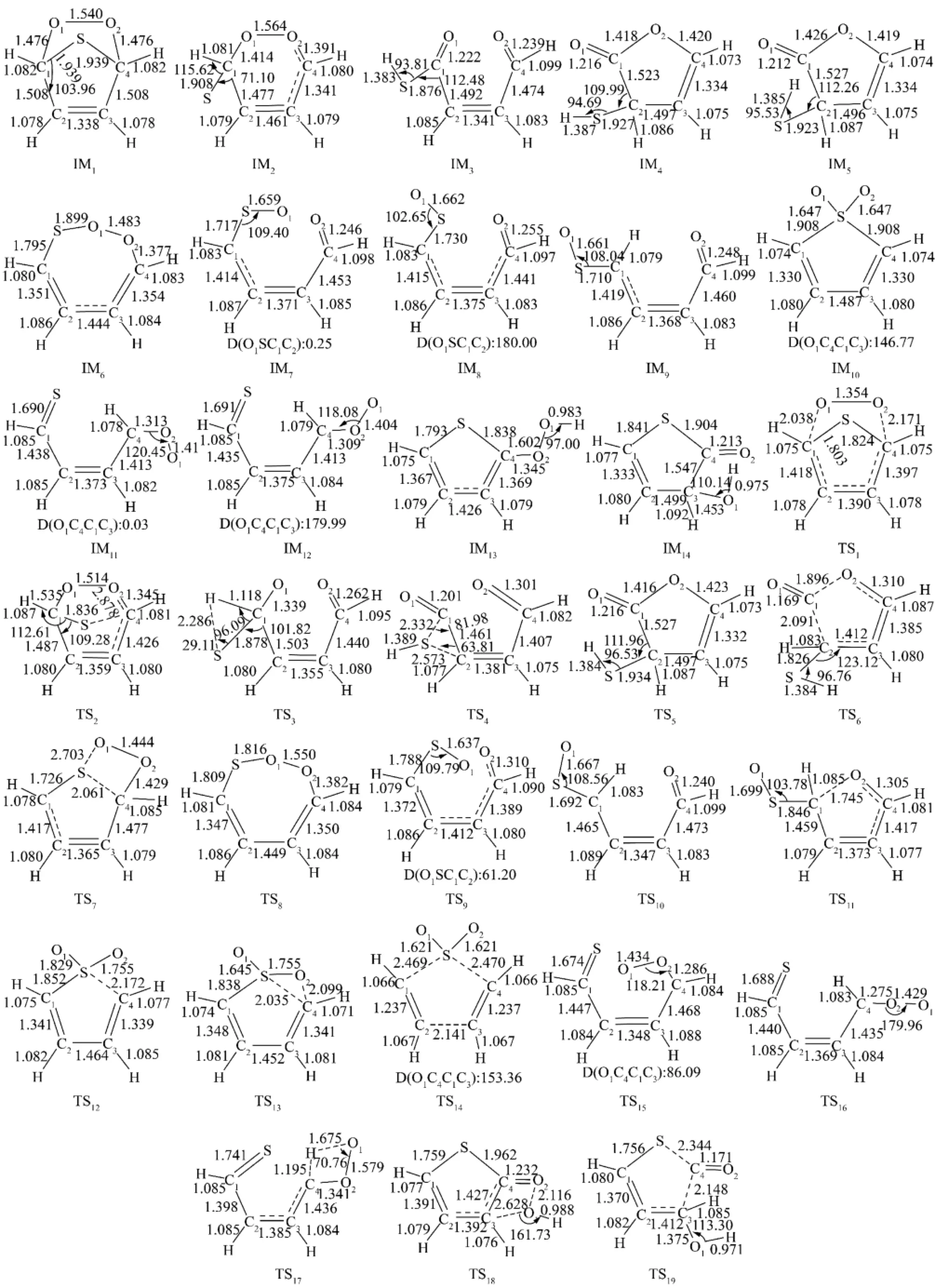
图2 在B3LYP/6-311G水平下计算中间体和过渡态的几何构型(键长单位:10-10m,键角单位:(°))Fig.2 Geometries of intermediate and transition states at the B3LYP/6-311G level(Bond lengths:10-10meters,bond angles:degrees)

表1 噻吩与O2反应的反应物、产物、中间体和过渡态的总能量(TE)、零点振动能(ZPVE)以及相对能量(RE)Table 1 Total energies(TE),zero-point vibration energies(ZPVE),and relative energies(RE)ofreactants, products,intermediate,and transition states on the reaction for thiophene with O2kJ/mol

图3 在B3LYP/6-311G水平下噻吩结构与O2反应的反应势能面剖面图Fig.3 Schematic profile of the potential energy surface for thiophene structure and O2at the B3LYP/6-311G level
2.2 反应路径分析
(1)噻吩结构与O2反应生成产物P1。
Path 1:从图3可以看出,反应物R经过过渡态TS1生成稳定的中间体IM1,此过程具体分析见2.1节。从图2可以看出,中间体IM1的S—C4键拉长,经过过渡态TS2,然后S—C4键断开生成中间体IM2,中间体IM1的S—C4键的键长为1.939×10-10m,而过渡态TS2的S—C4键长达到2.878×10-10m,IM1→IM2需要克服的势垒为72.39 kJ/mol(图3)。中间体IM2上与C1原子相连的H原子发生C1—S邻位迁移,同时O1—O2键作用力减弱并断开,经过过渡态TS3形成中间体IM3,这一过程需要克服的势垒为72.95 kJ/mol。中间体IM3的O2原子的电子向C1原子靠近,同时S—C1键变弱,H—S基从C1原子向C2原子迁移,经过过渡态TS4生成中间体IM4,O2原子与C1原子连接使中间体IM4成为环状结构,IM3→IM4需要克服的势垒为90.80 kJ/mol。中间体IM4经过旋转过渡态TS5异构化为中间体IM5,这一过程需要克服的势垒为8.68 kJ/mol。中间体IM5的C1—O2键和C1—C2键同时拉长,经过过渡态TS6分解为产物P1(HSC2H2CHO+CO),由IM5→P1需要克服很高的势垒(195.61 kJ/mol),此步在反应路径的竞争中处于劣势。这条路径的速控步骤为过渡态TS6(势垒为195.61 kJ/mol)。
(2)噻吩结构与O2反应生成产物P2。
Path 2:反应物R经过过渡态TS7生成稳定的中间体IM6,此过程具体分析见2.1节反应物的初始连接。中间体IM6的O1—O2键拉长,经过过渡态TS8后断开,异构化为稳定的中间体IM7,中间体IM6的O1—O2键长为1.483×10-10m,过渡态TS8的O1—O2键长为1.550×10-10m,IM6→IM7过程需要克服的势垒为6.41 kJ/mol。中间体IM7的O1—S键以S—C1键为轴旋转,经过过渡态TS9异构化为中间体IM8,中间体IM7的二面角D(O1SC1C2)为0.25°,过渡态TS9的二面角D(O1SC1C2)为61.20°,中间体IM8的二面角D(O1SC1C2)为180.00°,由IM7→IM8需要克服的势垒为60.64 kJ/mol。中间体IM8的C1原子自旋经过过渡态TS10,形成中间体IM9,由IM8→IM9需要克服的势垒为57.02 kJ/mol。中间体IM9的O2原子开始向C1原子靠近,同时S—C1键略微拉长,通过过渡态TS11后,O2原子和C1原子连接成单键,而S—C1键断裂,分解为稳定的产物P2,中间体IM9的S—C1键长为1.710×10-10m,过渡态TS11的S—C1键长为1.846×10-10m,IM9→P2需要克服的势垒为117.06 kJ/mol。
Path 3和Path 2的不同点在于反应物R经过不同的过渡态形成中间体IM8,在Path 3中,R→IM8经过过渡态TS12,这一过程需要克服势垒为315.99 kJ/mol,具体分析见2.1节反应物的初始连接。由IM8→P2的过程与Path 2的过程相同,在此不再赘述,具体分析请见上文。
Path 2和Path 3生成相同的产物P2(C4H4O+ SO)。Path2的速控步骤为TS11(势垒为117.06 kJ/mol);Path 3的速控步骤为TS12(势垒为315.99 kJ/mol);Path 2的速控步骤反应能垒比Path 3的速控步骤反应能垒低198.93 kJ/mol,显然,Path 2在反应路径的竞争中优于Path 3;Path 2与Path 1相比,Path 2的速控步骤反应能垒比Path 1的速控步骤反应能垒低78.55 kJ/mol,因此,Path 2在反应路径的竞争中也优于Path 1。
(3)噻吩结构与O2反应生成产物P3。
Path 4的反应物R异构化为中间体IM8的过程与Path 2相同,具体分析请见Path 2。在Path 4中,中间体IM8的S原子与C4原子作用力加强并形成单键,同时O2原子从C4原子向S原子迁移,经过过渡态TS13形成中间体IM10,过渡态TS13的S—C4键长为2.035×10-10m,中间体IM10的S—C4键长为1.908×10-10m,这一过程需要克服的势垒相当的大(421.61 kJ/mol),在反应路径的竞争中必处劣势。中间体IM10的S原子与两个O原子相连接的作用力很强,而环状结构中C1—C2键和C3—C4键由于共轭键的作用作用力也很强,因此S—C1键、S—C2键和C2—C3键同时拉长,经过过渡态TS14分解为产物P3,IM10→P3需要克服的势垒为244.22 kJ/mol。
Path 5中反应物R到中间体IM8的过程和Path 3相同,而Path 5中中间体IM8到产物P3的过程与Path 4相同,在此不再分析,具体分析请见上文。
Path 4和Path 5生成相同的产物P3(2C2H2+ SO2)。Path 4的第1速控步骤为过渡态TS13(势垒为421.61 kJ/mol),第2速控步骤为过渡态TS14(势垒为244.22 kJ/mol);Path 5的第1速控步骤与Path 4相同,第2速控步骤为过渡态TS12(势垒为315.99 kJ/mol);从两条反应路径的第1速控步骤可以看出,它们在反应路径的竞争中同样处于劣势,更无力与Path 2和Path 1相比;Path 4的第2速控步骤反应能垒比Path 5的第2速控步骤反应能垒低71.77 kJ/mol,显然,Path 4优于Path 5。
(4)噻吩结构与O2反应生成产物P4。
Path 6的初始连接方式与Path 2相同,分析同上。Path 6中中间体IM6的S—O1键断裂,经过过渡态TS15异构化为中间体IM11,IM6→IM11需要克服的势垒为123.98 kJ/mol。中间体IM11经过一个旋转过渡态TS16异构化为中间体IM12,O1原子以O2原子为原点绕其旋转,异构化前二面角D(OC4C1C3)为0.03°,而异构化后中间体IM12的二面角D (OC4C1C3)为179.99°,这个过程需要克服的势垒为150.93 kJ/mol。中间体IM12上与C4原子相连的H原子的电子向O1原子靠近,经过过渡态TS17后,这个H原子与C4原子断开,与O1原子连接,在H原子向O1原子迁移的过程中,S原子与C4原子的作用力加强形成单键,形成更稳定的中间体IM13,IM12→IM13需要克服的势垒为117.89 kJ/mol。中间体IM13经过H—O1基从O2原子迁移到C3原子,形成更稳定的中间体IM14,IM13→IM14经过过渡态TS18,这个过程需要克服的势垒为82.24 kJ/mol。中间体IM14的O2—C4键是一条双键,O2原子和C4原子相互作用力很强,又因为处在不断释放能量的环境中,S—C4和C3—C4键拉长,经过过渡态TS19,两条键断裂分解为产物P4,IM14→P4需要克服的势垒为124.17 kJ/mol。
Path 6生成产物P4(HOC2H2CHS+CO),这条路径的速控步骤为过渡态TS16(势垒为150.93 kJ/ mol);与Path 1相比,Path 6速控步骤的反应能垒比Path 1速控步骤的低44.68 kJ/mol;而比生成P2的Path 2高33.87 kJ/mol;因此,Path 6在反应路径中的竞争力强于Path 1,Path 2的竞争力优于Path 6。
综上所述,噻吩结构和O2反应的6条反应路径的难易程度为:Path 2>Path 6>Path 1>Path 3>Path 4>Path 5。
3 结 论
(1)经计算可知,噻吩结构与O2反应的6条反应路径中,路径Path 2(R→TS7→IM6→TS8→IM7→TS9→IM8→TS10→IM9→TS11→P2)的反应势垒最低,为主反应路径,其次就是路径Path 6(R→TS7→IM6→TS15→IM11→TS16→IM12→TS17→IM13→TS18→IM14→TS19→P4)。
(2)计算结果表明,噻吩结构与O2反应的4种产物中,产物P2(C4H4O+SO)是反应的主产物, P3(2C2H2+SO2)是最难生成的产物。
(3)煤结构中噻吩结构无需从外界吸收热量,与氧气接触即可被氧化。
[1] Gryglewicz G,Jasieńko S.Sulfur groups in the cokes obtained from coals of different ranks[J].Fuel Process Technol.,1988,19(1): 51-59.
[2] Miura K,Mae K,Shimada M,et al.Analysis of formation rates of sulfur-containing gases during the pyrolysis of various coals[J].Energ.Fuel,2001,15(3):629-636.
[3] 孙成功,李保庆,Snape C E.煤中有机硫形态结构和热解过程硫变迁特性的研究[J].燃料化学学报,1997,25(4):358-362.
Sun Chenggong,Li Baoqing,Snape C E.Characterization of organic sulfur forms in some chinese coals by high pressure TPR and sulphur transfer during hydropyrolysis[J].Journal of Fuel Chemistry and Technology,1997,25(4):358-362.
[4] 谢建军,杨学民,吕雪松,等.煤热解过程中硫氮分配及迁移规律研究进展[J].化工进展,2004,23(11):1214-1218.
Xie Jianjun,Yang Xuemin,Lü Xuesong,et al.Progress on transformation behavior of sulfur and nitrogen during coal pyrolysis[J].Chemical Industry and Engineering Progress,2004,23(11):1214-1218.
[5] Ling Lixia,Zhang Riguang,Wang Baojun,et al.Density functional theory study on the pyrolysis mechanism of thiophene in coal[J].Journal of Molecular Structure:THEOCHEM,2009,905(1):8-12.[6] Ling Lixia,Zhang Riguang,Wang Baojun,et al.DFT study on the sulfur migration during benzenethiol pyrolysis in coal[J].Journal of Molecular Structure:THEOCHEM,2010,952(1):31-35.
[7] 邓存宝,王继仁,邓汉忠,等.氧在煤表面—CH2—NH2基团上的化学吸附[J].煤炭学报,2009,34(9):1234-1238.
Deng Cunbao,Wang Jiren,Deng Hanzhong,et al.Chemical adsorption of O2adsorbed in the coal surface—CH2—NH2group[J].Jounal of China Coal Society,2009,34(9):1234-1238.
[8] 邓存宝,王雪峰,王继仁,等.煤表面含S侧链基团对氧分子的物理吸附机理[J].煤炭学报,2008,33(5):556-560.
Deng Cunbao,Wang Xuefeng,Wang Jiren,et al.Physical adsorption mechanism of coal surface containing sulfur group adsorption to more oxygen molecule[J].Jounal of China Coal Society,2008,33(5): 556-560.
[9] 邓存宝,王继仁,叶 兵,等.煤表面对单氧分子的物理吸附机理[J].中国矿业大学学报,2008,37(2):171-175.
Deng Cunbao,Wang Jiren,Ye Bing,et al.Physical mechanism of a single oxygen molecule adsorbs to the coal surface[J].Journal of China University of Mining&Technology,2008,37(2):171-175.
[10] 亓 雪,石秋杰,谌伟庆,等.Mo对非晶态合金Ni-B/薄水铝石催化剂上噻吩加氢脱硫性能的影响[J].催化学报,2012,33 (3):543-549.
Qi Xue,Shi Qiujie,Chen Weiqing,et al.Effect of Mo on performance of Ni-B/boehmite amorphous alloy catalyst for thiophene hydrodesulfurization[J].Chinese Journal of Catalysis,2012,33 (3):543-549.
[11] 高晓明,付 峰,武玉飞,等.Co-BiVO4光催化剂的制备及其用于光催化氧化噻吩[J].无机材料学报,2012,27(10):1073-1078.
Gao Xiaoming,Fu Feng,Wu Yufei,et al.Preparation of Co-BiVO4photocatalyst and its application in the photocatalytic oxidative thiophene[J].Journal of Inorganic Materials,2012,27(10):1073-1078.
[12] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 03w,version6.0,Gaussian[M].Inc.,Wallingford,CT,2004.
[13] Govind N,Petersen M,Fitzgerald G,et al.A generalized synchronous transit method for transition state location[J].Computational Materials Science,2003,28(2):250-258.
[14] Malick D K,Petersson G A,Montgomery Jr J A.Transition states for chemical reactions I.Geometry and classical barrier height[J].The Journal of Chemical Physics,1998,108(14):5704-5713.
[15] Bakken V,Helgaker T.The efficient optimization of molecular geometries using redundant internal coordinates[J].The Journal of Chemical Physics,2002,117(20):9160-9174.
[16] Ayala P Y,Schlegel H B.A combined method for determining reaction paths,minima,and transition state geometries[J].The Journal of Chemical Physics,1997,107(2):375-384.
Theoretical study on the mechanism of the thiophene structure with O2reaction
DAI Feng-wei1,DENG Cun-bao1,DENG Han-zhong2,WANG Xue-feng1,GAO Fei1,ZHANG Xun3
(1.College of Safety Science and Engineering,Liaoning Technical University,Fuxin 123000,China;2.College of Materials Science and Engineering,Liaoning Technical University,Fuxin 123000,China;3.College of Mining Engineering,Liaoning Technical University,Fuxin 123000,China)
In order to study the reaction mechanism of coal spontaneous combustion,the calculations were performed with the Gaussian 03 program.At the B3LYP/6-311G level,the reaction mechanism of thiophene type organic sulfur in coal structure with O2was studied by using density functional theory(DFT)method.The calculations show that initial connection of the reactant has three forms,the reactant of path 1 in it is readily isomerized to the intermediate IM1through the transition state TS1,this process is exothermic process without overcome potential barrier,it show that thiophene type organic sulfur in coal contact with oxygen to be oxidized without absorbing heat from outside;this reaction has six reaction paths,the path 2 is the main reaction path,its rate-controlling step is the transition state TS6,its reaction energy barrier is 117.06 kJ/mol,P2(C4H4O+SO)is the main product.
thiophene;reaction mechanism;Gaussian 03 program;density functional theory
TQ530
A
0253-9993(2014)04-0699-06
戴凤威,邓存宝,邓汉忠,等.噻吩结构与O2反应机理的理论研究[J].煤炭学报,2014,39(4):699-704.
10.13225/j.cnki.jccs.2013.0562
Dai Fengwei,Deng Cunbao,Deng Hanzhong,et al.Theoretical study on the mechanism of the thiophene structure with O2reaction[J].Journal of China Coal Society,2014,39(4):699-704.doi:10.13225/j.cnki.jccs.2013.0562
2013-05-02 责任编辑:毕永华
国家自然科学基金面上资助项目(51274112);国家自然科学基金资助项目(51174108)
戴凤威(1987—),女,辽宁辽阳人,博士研究生。Tel:0418-3351702,E-mail:daifengwei214@163.com
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
英语文摘(2020年7期)2020-09-21
山东工业技术(2018年9期)2018-05-26
今日财富(2017年32期)2017-10-19
中学化学(2017年5期)2017-07-07
中学化学(2016年4期)2016-05-30
股市动态分析(2015年12期)2015-09-10
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
中学化学(2014年1期)2014-04-23
中学生数理化·高二版(2008年5期)2008-11-12
