
环境胁迫诱导的细胞适应性突变
2014-05-25 00:32:49朱林江李崎
遗传 2014年4期
朱林江, 李崎
江南大学食品安全与营养协同创新中心, 教育部工业生物技术重点实验室, 无锡 214122
环境胁迫诱导的细胞适应性突变
朱林江, 李崎
江南大学食品安全与营养协同创新中心, 教育部工业生物技术重点实验室, 无锡 214122
细胞具有普遍的突变和进化能力, 如病原菌的抗药性、工业菌株的适应性和人体细胞的癌变等, 但是细胞的适应性突变是如何产生的呢?通过非致死性突变分析模型的建立与应用, 产生了新的适应性进化观点, 即环境胁迫诱导细胞适应性突变。这种环境诱导的细胞突变过程涉及多方面的生理调控, 包括细胞内毒性物质(如氧活性物质)积累并造成DNA损伤、DNA错配修复的活性受到抑制、胞内RpoS反应和SOS反应被激活等。这些反应使胞内高保真的DNA复制状态转变为低保真的DNA修复状态, 提高胞内突变率和重组活性。此外,基因转录影响基因组的不稳定, 容易产生DNA损伤, 并造成局部的高突变率, 即形成了转录偶联的DNA修复与突变为基础的适应性突变观点。文章围绕环境胁迫诱导细胞突变率增加和转录偶联的DNA修复与突变这两种适应性突变分子机制, 阐述其相关的研究进展, 以期更好地理解环境条件诱导细胞发生适应性突变的过程。
压力诱导突变; 适应性进化; 超突变态; 转录偶联的DNA突变
传统的达尔文进化理论认为, 生物进化是生物个体发生变异后, 通过自然条件选择的结果。生物的这种变异进化现象普遍存在, 如:临床上频繁出现的病原菌抗药性突变, 通过连续传代获得适应性进化的工业菌株, 人体细胞的癌变化以及免疫系统细胞内诱发产生新的抗体基因等。但是, 这些细胞突变是如何产生的?细胞的突变与外界的选择环境是否有关?细胞的突变率是固定不变的、小几率的,还是会被诱导增加的?这些问题涉及生物进化的动力和生物进化论的核心问题, 目前仍未能被清晰解答[1]。早在20世纪40、50年代, Luria等[2]和Lederberg等[3]利用 E.coli获得噬菌体抗性和抗生素抗性的突变模型, 并经过数学统计分析, 证明细胞的随机突变速率是个常数, 与环境无关, 为以随机突变为基础的进化理论奠定坚实的基础, 从而形成以DNA复制过程中产生的随机突变为基础的新达尔文进化论(neo-Darwinism)的主流观点。不过, 上述实验模型采用了致死性的选择条件, 这种选择性条件比较剧烈,其阳性突变主要来自于细胞分裂过程中的随机突变。后来发展出非致死性选择条件的突变分析模型,包括细菌的研究模型[4~6]、新发展的酵母研究模型[7]以及国内学者构建的细菌分析模型[8,9]。这些突变模型提高了突变分析的灵敏度, 能够分析突变产生的胞内生理过程, 并形成了压力诱导细胞适应性突变的新进化观点[1,10~12], 即胁迫条件诱导细胞的生理变化, 使细胞的突变率增加, 产生易于突变的状态,促进适应性突变的产生。
尽管胁迫条件能够诱导多种不同类型的基因突变, 包括少量碱基的改变、删除或插入、基因拷贝数的改变、可移动元件发生移动以及大片段的基因组重排等, 但是适应性突变的产生过程涉及了一些共同的分子机制[1]。本文总结了一些压力条件诱导细胞突变的分析模型; 分析压力诱导细胞突变的相关分子机制, 强调胁迫环境诱导胞内高突变状态的产生; 结合转录偶联的DNA突变和修复机制, 形成环境条件诱导细胞适应性突变的分子机制。
1 非致死性选择条件的突变分析模型
当细胞处于营养匮乏等非致死性胁迫环境条件时, 细胞处于缓慢生长或静止状态, 但是细胞保持活性, 此时胞内的突变率被诱导增加, 并易于产生适应性突变。当在胁迫条件中培养时间的延长, 适应性突变的细胞数量会不断增加, 如 Lac-表型的回复突变、氨基酸营养缺陷型的回复突变和老化菌落中提高的突变能力等。
最早报道的适应性突变分析模型是 Shapiro的E.coli系统[4]。该菌株携带 Mu噬菌体, 其插入到araB基因(L-核酮糖激酶)中, 抑制下游基因 lacZ和lacY的表达, 使菌株表现为乳糖利用能力缺陷型(Lac-)。若将108个细胞涂布到乳糖的最小培养基表面, 在 3周培养过程中不断出现生长的菌落, 并布满整个平板。若培养基中不含乳糖, 在 3周培养过程中细胞不会死亡, 但不能形成 AraB-LacZ+的突变菌落, 证明乳糖饥饿环境诱导发生适应性突变。所以该实验模型证实乳糖饥饿环境偏好性地诱导噬菌体Mu删除突变, 形成araB-lacZ融合基因, 并通过araB的启动子进行表达, 获得Lac+的突变表型。而非诱导性环境中或正常富集培养基中, Mu在基因组中稳定存在, 其删除突变率几乎不可测。同样的情况发生在另一株 E.coli菌中, 乳糖的饥饿胁迫同样偏好性地诱导基因组上 ebg操纵子的突变, 从而积累 Lac+的表型[13]。除了乳糖饥饿胁迫外, 氨基酸的饥饿胁迫同样偏好性地诱导细胞适应性突变的产生[12]。当E.coli菌株色氨酸合成酶的α或β亚基发生突变, 表现为色氨酸的营养缺陷型(Trp-)。在正常培养条件下, 菌株不能回复突变产生 Trp+的表型。当细胞涂布于添加少量色氨酸的最小培养基上时, 经过10 d的培养, 不断有Trp+表型的菌落被诱导产生。所以, 色氨酸饥饿胁迫同样偏好性地诱导色氨酸合成酶亚基的回复突变, 得到适应性突变的表型。
以上乳糖回复突变和色氨酸回复突变实验证实胁迫环境提升细胞的适应性突变。在自然环境条件下, 微生物也总是处于营养匮乏的条件, 那么天然的胁迫环境是否诱导细胞提升适应性突变呢?天然分离菌株的突变能力是否与环境相关?对此, Bjedov等[14]从世界范围(包括法国、墨西哥、委内瑞拉、马里、澳大利亚、克罗地亚以及南极)人或动物的粪便、肠道中分离出787株天然E.coli菌株, 比较在固体培养基上形成老化菌落(即接近自然条件的营养匮乏状态)后的突变率差异。通过比较培养 1 d的新生菌落和 7 d的老化菌落的利福平抗药性突变率, 结果表明 7 d老化菌落的利福平抗药性突变率显著提高, 787株菌的老化菌落的突变率平均提高10倍, 其中13%的菌株突变率提高大于100倍[14]。这种老化菌落的突变率增加与细胞受饥饿胁迫密切相关。而不同环境中分离的菌株的突变率差异与它们的生存环境密切相关, 而与系统发育树上的亲缘关系无关。同样, 对天然分离的幽门螺杆菌(Helicobacter pylori)的突变能力分析表明[15], 其典型的高突变率遗传背景与其胁迫性的生存环境相关, 其中高浓度的氧活性物质或氮活性物质均显著提高了基因突变率或遗传重组活性。因此, 生存的环境条件与天然菌株的突变能力密切相关。
在众多非致死性选择条件的突变模型中, 20世纪80年代末Cairns和Foster开发的lac回复突变系统, 即Cairns系统, 被研究得最详细[1,16]。该模型的模式菌株为 E.coli FC40, 其基因组上已经突变删除lac操作子, 引入的F′质粒上携带lacI-lacZ的融合基因lacIΩZ, 该融合基因中间位置发生+1读码框突变,所以菌株表现为Lac-。这个融合基因在突变的Iq启动子下组成型表达。正常条件下, 表达的融合蛋白具有1%~2%的β-半乳糖苷酶活性。该菌株在乳糖饥饿胁迫下, 可偏好性地诱导lacIΩZ读码框突变的回复突变或该融合基因的基因扩增(50~100倍), 使Lac+菌落不断出现。Lac+菌落数的增加与培养时间大致呈线性关系。这些回复突变菌落的基因型主要分为两类:(1) 点突变, 即 lacIΩZ基因内部发生点突变(包括缺失和插入), 回复形成正确的读码框; (2)基因扩增, 包含 lacIΩZ基因的片段单元(大小不一)在细胞内 50~100倍的扩增, 从而过量表达具有1%~2%的 β-半乳糖苷酶活性的融合蛋白, 恢复细胞利用乳糖的能力[17]。这些回复突变的产生, 只有在乳糖为唯一碳源的饥饿胁迫条件时才会发生, 证明了乳糖饥饿条件的诱导作用。由于Cairns模型能够灵敏地定量分析细胞的适应性突变过程, 所以被广泛地用于研究压力诱导细胞适应性突变的分子机制。
2 压力诱导细胞突变的相关分子机制
微生物突变实验表明非致死性的选择条件能够偏好性地诱导相关基因发生适应性突变, 而且这种遗传突变发生在缓慢生长的细胞中[12,18]。随着培养时间的延长, 突变细胞的数量不断增加, 如 Cairns系统的 Lac+回复突变率大致呈线性增长的趋势[6]。所以, 压力诱导细胞突变与DNA复制过程产生的随机突变存在显著的差异, 前者的适应性突变率提高102~103倍。研究表明, 多个分子机制涉及压力诱导细胞突变的过程[10,19~21], 如图1所示。
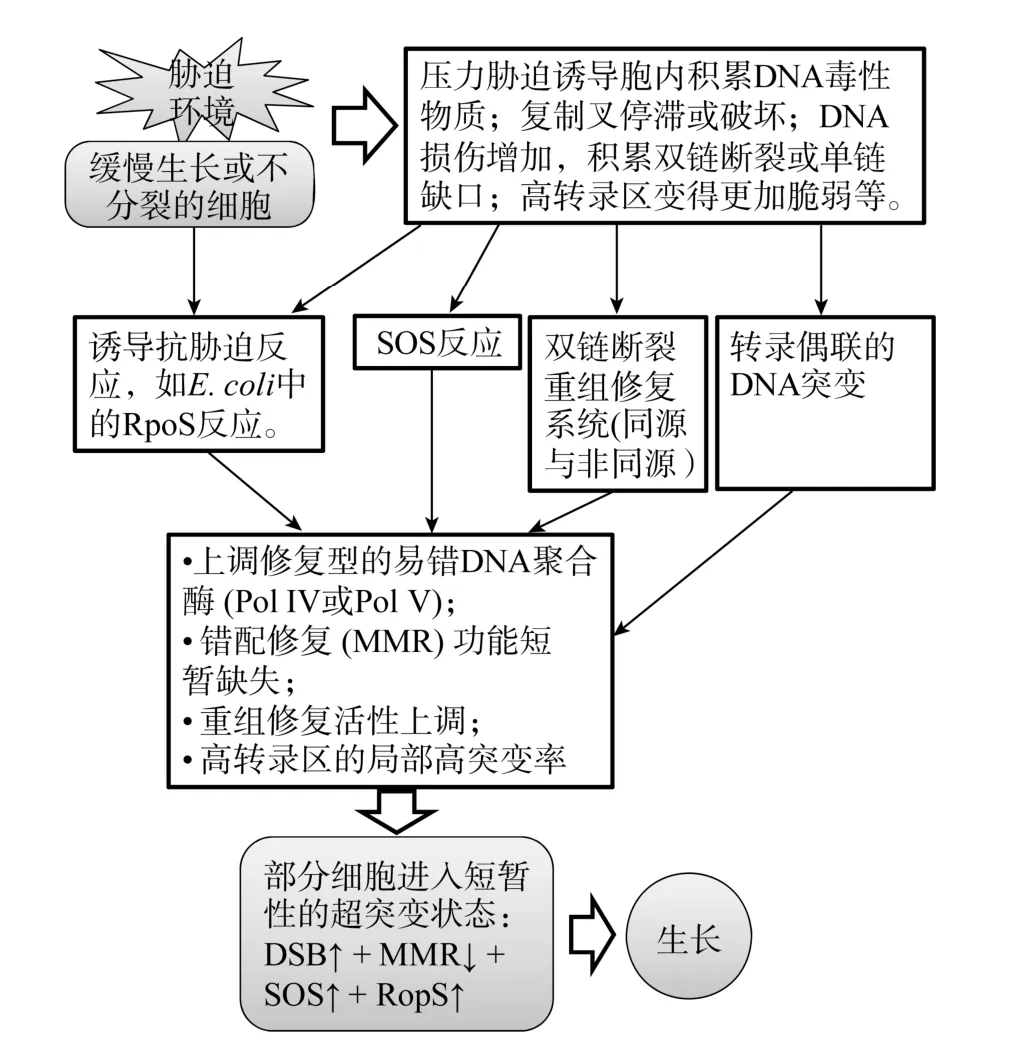
图1 压力诱导细胞适应性突变的分子机制当细胞受到环境胁迫, 胞内积累有毒物质而造成 DNA损失, 如基因组上的单链缺口或双链断链(Double-strand break, DSB), 诱导了胞内抗胁迫反应、SOS反应和高活性的DNA重组反应[10,21],导致细胞的突变率增加[22]; 环境诱导的高活性基因转录增加该区域基因组的不稳定性, 表现为转录区的超突变特征[19,20]; 这些生理反应的共同累积, 使细胞形成超突变态, 少量细胞获得适应性突变而具备生长能力。
2.1 胞内有毒物质的积累和损伤DNA的积累
当细胞处于正常条件时, 胞内代谢迅速, DNA复制高保真, 细胞保持较低的突变率。当细胞处于胁迫环境时, 胞内代谢受到抑制, 容易积累有毒物质, 如氧活性物质(Reactive oxygen species, ROS)和被修饰的核苷酸, 造成 DNA的损伤; 积累了 DNA单链缺口或双链断裂, 诱导细胞的生理状态变化。胞内有毒物质和损伤 DNA的积累被认为是压力诱导细胞突变的起始阶段[10,21], 如图1所示。如Bjedov等[14]分析表明, 自然界分离的E. coli菌株在菌落老化阶段的突变率显著提高, 与有氧培养条件密切相关。老化菌落中的细胞代谢活性较弱, 在有氧条件下, 胞内容易积累ROS, 增加DNA的损伤, 从而提高细胞的突变率。针对抗生素胁迫条件下细胞如何死亡的最新研究结果也证实了这一观点。当胞内代谢受到致死性的抗生素浓度阻碍时, 胞内积累大量的ROS和氧化的核苷酸, 使DNA损伤增加, 在修复型DNA聚合酶Pol Ⅳ作用后, 产生了大量的DNA双链断裂并导致细胞死亡[23]。但若降低抗生素的浓度, 即亚致死浓度的胁迫条件下, ROS对DNA的损伤程度减弱, 则诱导细胞发生多种抗药性突变[24]。而在Cairns模型系统中, Lac+回复突变率与F′质粒上能够产生由内切酶TraⅠ介导的单链DNA缺口密切相关; 若删除traI基因, Lac+回复突变率降低130倍[25]。若在lacIΩZ基因上游或下游人工引入可诱导的双链断裂位点, 能够有效地提高 Lac+回复突变率约 50倍[25], 这种诱导性同样在染色体基因组中适用[26]。所以, 当细胞处于胁迫环境中时, 胞内有毒物质的积累和损伤DNA的积累成为直接因素, 诱导胞内突变率的提高, 促进适应性突变的产生[10,21]。
2.2 限制性修复系统MMR的调控
胁迫条件诱导细胞适应性突变的过程中, MMR (Mismatch repair)活性的调控起到重要的作用。MMR是进化上非常保守的DNA错配修复系统, 能够校正DNA复制过程中产生的错配碱基, 提高 DNA复制的保真性近1000倍[27]。若MMR活性缺失, 细胞表现出高突变和高重组的表型, 其随机突变率提高近100倍; 胞内的一些微卫星重复序列非常不稳定, 易于重组, 且胞内基因组上易于整合外源的DNA[27,28]。MMR系统主要包括3个核心蛋白, MutS、MutL和MutH。MutS识别错配碱基, 并募集 MutL, 形成的复合物激活核酸内切酶MutH, 同时MutL募集解螺旋酶, 复合体共同作用降解未甲基化的新生的错配DNA链, 形成的DNA缺口由DNA聚合酶和连接酶完成修复[28]。在对数期的细胞中, MMR活性正常表达, 保证细胞处于高保真的DNA合成状态和较低的随机突变率。当进入稳定期或细胞处于胁迫环境下,胞内的MMR活性被下调, 其中关键蛋白MutL功能受到限制, 细胞的突变率显著增加[29]。所以, MMR活性的调控与胞内基因组突变率的变化密切相关。以 Cairns系统为例, 若组成型缺失 MMR活性, 使Lac+适应性回复突变率提高 100倍; 若人为上调MMR活性, 则显著降低Lac+突变率[29,30]。此外, 在天然菌株的适应性进化上, MMR活性的调控也具有重要意义。MMR组成型缺失的天然菌株也经常被分离得到, 表现出更好的适应性进化能力[31], 如上文提及的幽门螺杆菌[15]。通过系统发育树比较分析许多天然菌株的 MMR, 结果表明这些基因具有典型的镶嵌结构, 这可能源于高频率基因水平转移的结果[32]。本实验室的研究结果表明, MMR功能被组成型突变的菌株, 能更迅速地适应高渗透压、高温和高丁醇的胁迫环境[33]。所以, MMR活性的反复缺失与获得, 及其在胞内的调控, 在生物进化中可能起到关键作用[32]。
2.3 抵抗环境胁迫的胞内生理反应
细胞往往通过改变转录程序、改变自身的生理状态适应不同的环境条件。如在E.coli中存在7个σ因子, 结合RNA聚合酶, 识别不同类型基因的启动子, 根据外界环境决定基因的表达。其中σ70或RpoD在对数生长时期发挥作用, 并控制大部分看家基因的表达。当细胞进入稳定期或处于胁迫环境下, RpoD 将被 RpoS(σ38)取代, 激活压力反应(General stress response), 直接或间接地调控近340个基因的表达(包括关闭合成代谢的基因), 上调抗胁迫反应相关的基因表达, 用以抵抗胞外的营养缺乏、热胁迫、渗透压胁迫、不适pH胁迫以及氧化胁迫等[34]。此外, RpoS反应也参与胞内突变率的调控, 当缺失RpoS反应时, Cairns系统中的Lac+回复突变率降低10倍[35,36]。作为转录调控因子, RpoS蛋白不能直接参与基因突变过程, 推测其调控的一些蛋白参与了基因突变的产生过程, 如修复型的易错DNA聚合酶IV(DinB)。RpoS反应能够诱导并提高DinB的合成约2倍[35]。DinB是一种易错DNA聚合酶, 被认为直接参与 Lac+的适应性点突变, 这是由于 lac适应性点突变一般是在lacIΩZ基因内部发生-1读码框的回复突变, 而这种突变类型恰好是DinB的偏好性突变产物[37,38]。但是, 也有研究认为DinB与适应性突变无关[39], 且一些菌株中不含该类型的 DNA聚合酶[15]。此外, RpoS反应的上调提高了DNA双链断裂处的重组修复突变率[25]。
另一个被证明参与Lac+适应性回复突变的σ因子是σE, 即RpoE蛋白。RpoE反应属于外膜应激反应, 如外膜蛋白失活或折叠错误等, 控制胞内近200个基因的表达。大部分基因涉及脂多糖的合成、组装和动态变化等, 在维持细胞完整性上发挥重要功能; 其余一部分基因涉及蛋白质的转录和翻译, 以及DNA的合成和修复[40]。最近的研究表明, 通过转座子插入, 使σE失活, FC40菌株的Lac+回复突变率减少 11~15倍, 其中 Lac+回复突变的基因扩增型突变子减少近300倍, 即σE的失活严重抑制胞内基因扩增的突变产生[41]。σE被认为参与两种分子机制:(1)σE的缺失, 降低核酸内切酶 TraⅠ的表达水平, 从而减少 F′质粒产生 TraⅠ依赖的单链缺口, 使 Lac+回复突变率降低。通过过量表达 I-SceⅠ, 诱导细胞产生DSB, 能够补偿σE的缺失, 恢复约80%的突变率; (2)σE的缺失严重降低基因扩增的突变基因型, 表明σE以一种未知机制影响lacIΩZ等位基因的扩增[41]。
2.4 胞内SOS反应
胞内积累过量损伤的DNA时, 激活胞内的SOS反应, 提高DNA修复能力。其过程是RecA与损伤的单链DNA(Single-strand DNA, ssDNA)形成复合物,降解阻遏蛋白LexA, 诱导SOS反应, 上调近40个基因表达。这些基因涉及DNA修复、细胞分裂控制、跨损伤合成的DNA聚合酶等[42]。如E. coli中, Y-家族两个典型的修复型 DNA聚合酶 Pol IV(DinB)和PolⅤ(UmuD′2C)以及PolⅡ均是受 SOS反应调控。从细菌到动物, Y-家族的DNA聚合酶都是非常保守的, 具有跨损伤合成DNA的能力, 增强细胞对DNA损伤的耐受性。但是, 当它们作用于正常的DNA模板时, 易于产生突变, 不具有校正功能, 所以被称为易错DNA聚合酶[43,44], 与突变的产生密切相关。一些菌株的抗生素抗性突变依赖于SOS反应。若抑制胞内SOS反应, 则会减弱细胞的抗生素抗性突变,这种SOS介导的突变可能涉及易错DNA聚合酶的调控[45]。SOS反应增加突变率可能由于高活性低保真的DNA修复和重组。SOS反应上调近10倍的DinB。当DinB作用于完好的DNA模板时, 易于产生-1读码框突变。在对数期生长的细胞中, 这种突变容易被 MMR系统修复。而稳定期或受环境胁迫的细胞中, MMR功能则受到限制, 所以容易产生可遗传的基因突变。若删除 dinB基因, Lac+回复突变细胞率则降低至原来的 25%[46,47]。胞内 RecA依赖的同源重组功能是发生压力诱导的Lac+回复突变或环丙沙星诱导的细胞突变所必须 的[6,45]。在正常条件时,双链断裂的同源重组修复是高保真的。而当胞内的SOS反应被激活后, 过量上调的易错DNA聚合酶使同源重组修复趋向基因突变的产生。所以, SOS反应的激活提高胞内修复损失DNA的活性, 增加细胞对DNA损失耐受性, 同时提高了细胞的突变能力[1,11]。
3 压力诱导细胞生理状态的跃迁
采用以上描述的非致死性选择条件的突变分析模型表明, 细胞的基因突变的主要来源并非是细胞快速生长过程中的DNA复制错误, 而是源于缓慢生长的细胞自身诱发的适应性突变。其实, 这种缓慢生长状态更加接近于自然界微生物生存的状态, 而且被证明可用于适应性进化的菌种改良[33]。细胞的生长状态与突变率密切相关。当在富营养条件时,细胞快速生长, 胞内有毒化学物质积累的浓度较低,胞内保持较高的MMR活性, 实现高保真的DNA复制并合成子代DNA, 所以基因组的突变率微小。但是, 当细胞处于胁迫环境中, 如营养缺乏的稳定期阶段时, 细胞代谢和生长缓慢, 容易积累有毒化学物质, 造成大量的 DNA损失, 诱导 SOS反应和RpoS反应, 胞内的生理状态由原来高保真状态跃迁至低保真状态, 易产生突变[1,10]。因此, 压力诱导细胞突变的过程中, 涉及典型的突变状态跃迁, 如图2所示。

图 2 压力诱导细胞突变过程中涉及的胞内突变状态跃迁正常生长的细胞, 其代谢迅速, 不易积累过量的有毒物质, DNA损伤程度较低, 错配修复系统MMR活性正常, 细胞处于高保真的DNA合成状态。当细胞受到环境胁迫, 胞内容易积累毒性物质而造成DNA过度损伤, 诱导胞内的易于突变的生理状态[1,21,25]。
若E.coli胞内同时诱导激活RpoS反应和SOS反应, 细胞被认为处于超突变(Hypermutation)状态[1]。E.coli FC40在乳糖的M9平板上出现的大部分Lac+回复突变菌落被认为来源于超突变态细胞诱导过程。这种高突变能力除了表现在高 Lac+回复突变率外, 同时其二级非选择性的突变率比 Lac-细胞平均提高了约50倍, 这种提高的随机突变率被称为短暂的超突变状态[48,49]。Rosche等[49]研究表明, 这些超突变态细胞的出现概率约为 10-3, 而胞内的突变率比正常细胞提高约200倍。根据该结果, Foster[50]推算只有10%的Lac+回复突变细胞来自于超突变态。Torkelson等[48]则认为, Lac+回复突变细胞应全都来自于超突变态细胞, 产生超突变态细胞的概率应是10-4~10-3, 所以超突变态细胞的突变率应该提高104~106倍。尽管Roth等[51]提出反驳, 若胞内的突变率比正常细胞高105倍, 会导致基因组积累过多的无义突变而使细胞死亡, 但 Cairns等[52]则认为, 如果压力诱导突变率的提高集中在特殊的基因组区域,则突变率提高到正常条件的 105倍是可能的, 如E.coli FC40细胞发生Lac+回复突变时, F′质粒上的突变率高于基因组。目前这种超突变态假说仍缺乏直接的证据。另一种不同于超突变态假说的观点是基因扩增假说, 从基因扩增角度解释基因突变产生的过程[16,53]。不管基因突变产生于DNA修复过程还是基因扩增过程, 但是, 当细胞处于胁迫环境时,细胞的生理状态发生变化是必然发生的。这种状态跃迁提高了胞内DNA修复和重组活性, 同时增加了基因组突变率, 从而促进压力诱导的突变[22]。
4 转录偶联的DNA突变与修复
上述的非致死性突变模型中, 特殊基因的高突变率被偏好性地诱导, 这种适应性突变现象可能涉及转录偶联的DNA突变过程。当细胞处于特殊的胁迫环境时, 胞内诱导启动相关基因的转录和表达,同时关闭其他基因; 由于代谢受到抑制, 胞内积累过量的毒性物质, 容易造成DNA损伤; 基因转录增加 DNA的不稳定性[20], 如产生单链 DNA, 容易受到酶或其他毒性物质的攻击, 产生缺口; 转录过程与复制过程冲撞时, 在DNA链中容易渗入dUTP随后产生缺口[54]; 压力诱导的低保真的 DNA聚合酶过量积累, 细胞处于突变状态, 所以, 转录偶联的DNA突变机制使细胞在胁迫环境下提高基因组上应激表达的转录区的突变率, 造成基因组范围内突变率的非均一性分布, 表现为较高的适应性突变特征[55]。已有证据表明基因转录频率的提高, 基因突变率也随之提高。例如在酵母菌中, 移码突变基因lys2在诱导型启动子控制下分析其回复突变率, 结果表明回复突变率与基因转录频率具有显著相关性[56]。在E.coli中, 氨基酸合成途径中的酶(leuB)发生突变失活, 造成氨基酸营养缺陷型。若加速该突变基因的转录频率, 则能有效地提高该基因的回复突变率, 即 leuB-的回复突变率与细胞内 leuB-的mRNA水平成正比关系[57]。
转录偶联 DNA适应性突变的分子机制仍缺乏直接的证据。Wright[58]认为涉及转录过程诱导单链DNA形成颈环结构对突变率增加起到重要作用, 并通过计算预测, 颈环结构可提高基因的突变率。动物细胞的免疫球蛋白基因中, 转录偶联的基因组重组和突变也已被研究[59,60]。最近麻省理工学院的一个研究组发现转录延伸因子 NusA 的新功能[61~63],即当转录机器RNAP在延伸过程遇到缺口时, NusA能够富集跨损伤修复型DNA聚合酶DinB, 即PolⅥ。若消除NusA与DinB的相互作用, 压力诱导的 lac回复突变率显著降低。根据NusA在DNA修复过程中的新功能, 可将压力诱导突变理论与转录偶联的DNA修复与突变机制相结合, 建立基因转录与基因复制、修复和突变之间的关系, 使胞内的DNA代谢与RNA代谢之间相互联系[64], 从而形成了胁迫环境能够诱导的基因组范围的适应性突变理论, 但是该理论仍需要实验的直接证据。
5 结 语
早在150年前达尔文就提出自然选择的生物进化论, 认为生物进化是生物个体发生变异、自然选择的结果。20世纪40~50年代, 通过经典的随机突变实验确认细胞的基因突变出现在选择之前, 突变率是一个常数, 与选择环境无关, 形成新达尔文主义的观点。但是, 近年来, 改进突变分析模型, 并采用非致死性选择条件后, 细胞的突变率被证实不是一个常数, 而是一个变量, 而且环境的选择性条件可偏好性诱导适应性突变。所以, 外界的环境条件被认为能够诱导基因突变, 从而促进生物的进化。这种新的适应性进化论观点仍存在争议, 主要集中于基因突变产生的过程, 即基因突变是在什么时间、哪一步反应产生以及怎样的过程中产生等。随着胁迫环境诱导细胞突变过程的深入研究, 形成了细胞突变被诱导产生的观点; 即当细胞处于胁迫条件时, 细胞生长速率缓慢, 胞内容易积累有毒物质,从而导致胞内产生大量的损伤DNA; 这些有毒物质和损伤的DNA会激活胞内的压力胁迫反应和 SOS反应; 细胞的DNA合成由原来的高保真复制状态转变为易于突变的修复状态, 从而产生适应性突变。而针对胁迫环境偏好性诱导适应性突变产生的问题,本文提出将转录偶联的 DNA修复突变机制与压力诱导细胞突变状态跃迁相结合的适应性突变假说:即胁迫环境会诱导相关基因的高水平转录; 同时诱导高突变状态, 导致高转录区转变为高突变区, 使环境条件诱导的基因具有高突变率, 最终易于产生适应性突变, 释放细胞的压力, 恢复高保真的正常状态。但是这种假说仍需要进一步研究有关转录区域易于适应性突变的直接证据。
[1] Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol, 2007, 42(5): 399-435.
[2] Luria SE, Delbruck M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics, 1943, 28(6): 491-511.
[3] Lederberg J, Lederberg EM. Replica plating and indirect selection of bacterial mutants. J Bacteriol, 1952, 63(3): 399-406.
[4] Shapiro JA. Observations on the formation of clones containing araB-lacZ cistron fusions. Mol Gen Genet, 1984, 194(1-2): 79-90.
[5] Hall BG. Directed evolution of a bacterial operon. Bioessays, 1990, 12(11): 551-558.
[6] Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics, 1991, 128(4): 695-701.
[7] Shor E, Fox CA, Broach JR. The yeast environmental stress response regulates mutagenesis induced by proteotoxic stress. Plos Genet, 2013, 9(8): E1003680.
[8] 张汉波, 沙涛, 程立忠, 丁骅孙, 施雯, 于春蓓, 张会荣.大肠杆菌FC40系统静止期突变中的F因子转移. 遗传, 2003, 25(4): 428-432.
[9] 吕忠, 王敖全. 研究适应突变的一个新的实验系统. 中国科学C辑, 2000, 30(5): 547-553.
[10] Rosenberg SM. Evolving responsively: adaptive mutation. Nat Rev Genet, 2001, 2(7): 504-515.
[11] Foster PL. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol, 2007, 42(5): 373-397.
[12] Foster PL. Mechanisms of stationary phase mutation: a decade of adaptive mutation. Annu Rev Genet, 1999, 33: 57-88.
[13] Hall BG. Spectra of spontaneous growth-dependent and adaptive mutations at ebgR. J Bacteriol, 1999, 181(4): 1149-1155.
[14] Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. Stress-induced mutagenesis in bacteria. Science, 2003, 300(5624): 1404-1409.
[15] Kang JM, Iovine NM, Blaser MJ. A paradigm for direct stress-induced mutation in prokaryotes. FASEB J, 2006, 20(14): 2476-2485.
[16] Roth JR, Kugelberg E, Reams AB, Kofoid E, Andersson DI. Origin of mutations under selection: the adaptive mutation controversy. Annu Rev Microbiol, 2006, 60: 477-501.
[17] Hastings PJ, Bull HJ, Klump JR, Rosenberg SM. Adaptive amplification: an inducible chromosomal instability mechanism. Cell, 2000, 103(5): 723-731.
[18] 张汉波, 沙涛, 程立忠, 丁骅孙. 适应性突变的遗传学特征. 遗传, 2002, 24(3): 395-398.
[19] Burch LH, Yang Y, Sterling JF, Roberts SA, Chao FG, Xu H, Zhang LL, Walsh J, Resnick MA, Mieczkowski PA, Gordenin DA. Damage-induced localized hypermutability. Cell Cycle, 2011, 10(7): 1073-1085.
[20] Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nature Rev Gene, 2012, 13(3): 204-214.
[21] Rosenberg SM, Shee C, Frisch RL, Hastings PJ. Stressinduced mutation via DNA breaks in Escherichia coli: a molecular mechanism with implications for evolution and medicine. Bioessays, 2012, 34(10): 885-892.
[22] Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. Impact of a stress-inducible switch to mutagenicrepair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci USA, 2011, 108(33): 13659-13664.
[23] Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science, 2012, 336(6079): 315-319.
[24] Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radicalinduced mutagenesis. Mol Cell, 2010, 37(3): 311-320.
[25] Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell, 2005, 19(6): 791-804.
[26] Shee C, Gibson JL, Rosenberg SM. Two mechanisms produce mutation hotspots at DNA breaks in Escherichia coli. Cell Rep, 2012, 2(4): 714-721.
[27] Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol, 2003, 57: 579-608.
[28] Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem, 2005, 74: 681-710.
[29] Harris RS, Feng G, Ross KJ, Sidhu R, Thulin C, Longerich S, Szigety SK, Winkler ME, Rosenberg SM. Mismatch repair protein MutL becomes limiting during stationaryphase mutation. Genes Dev, 1997, 11(18): 2426-2437.
[30] Foster PL, Cairns J. Mechanisms of directed mutation. Genetics, 1992, 131(4): 783-789.
[31] LeClerc JE, Li B, Payne WL, Cebula TA. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science, 1996, 274(5290): 1208-1211.
[32] Denamur E, Lecointre G, Darlu P, Tenaillon O, Acquaviva C, Sayada C, Sunjevaric I, Rothstein R, Elion J, Taddei F, Radman M, Matic I. Evolutionary implications of the frequent horizontal transfer of mismatch repair genes. Cell, 2000, 103(5): 711-721.
[33] Zhu L, Cai Z, Zhang Y, Li Y. Engineering stress tolerance of Escherichia coli by stress-induced-mutagenesis (SIM) based adaptive evolution. Biotechnol J, 2013, doi: 10. 1002/biot. 201300277.
[34] Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev, 2002, 66(3): 373-395.
[35] Layton JC, Foster PL. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol, 2003, 50(2): 549-561.
[36] Lombardo MJ, Aponyi I, Rosenberg SM. General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics, 2004, 166(2): 669- 680.
[37] Rosenberg SM, Longerich S, Gee P, Harris RS. Adaptive mutation by deletions in small mononucleotide repeats. Science, 1994, 265(5170): 405-407.
[38] Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc Natl Acad Sci USA, 1997, 94(25): 13792-13797.
[39] Koskiniemi S, Hughes D, Andersson DI. Effect of translesion DNA polymerases, endonucleases and RpoS on mutation rates in Salmonella typhimurium. Genetics, 2010, 185(3): 783-795.
[40] Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. Conserved and variable functions of the sigmaE stress response in related genomes. PLoS Biol, 2006, 4(1): E2.
[41] Gibson JL, Lombardo MJ, Thornton PC, Hu KH, Galhardo RS, Beadle B, Habib A, Magner DB, Frost LS, Herman C, Hastings PJ, Rosenberg SM. The sigma(E) stress response is required for stress-induced mutation and amplification in Escherichia coli. Mol Microbiol, 2010, 77(2): 415-430.
[42] Courcelle J, Khodursky A, Peter B, Brown PO, Hanawalt PC. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics, 2001, 158(1): 41-64.
[43] Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu Rev Microbiol, 2006, 60: 231-253.
[44] Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol, 2012, 13(3): 141-152.
[45] Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol, 2005, 3(6): E176.
[46] Foster PL. Adaptive mutation in Escherichia coli. Cold Spring Harb Symp Quant Biol, 2000, 65: 21-29.
[47] McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell, 2001, 7(3): 571-579.
[48] Torkelson J, Harris RS, Lombardo MJ, Nagendran J, Thulin C, Rosenberg SM. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J, 1997, 16(11): 3303-3311.
[49] Rosche WA, Foster PL. The role of transient hypermutators in adaptive mutation in Escherichia coli. Proc Natl Acad Sci USA, 1999, 96(12): 6862-6867.
[50] Foster PL. Adaptive mutation in Escherichia coli. J Bacteriol, 2004, 186(15): 4846-4852.
[51] Roth JR, Kofoid E, Roth FP, Berg OG, Seger J, Andersson DI. Regulating general mutation rates: examination of the hypermutable state model for Cairnsian adaptive mutation. Genetics, 2003, 163(4): 1483-1496.
[52] Cairns J, Foster PL. The risk of lethals for hypermutating bacteria in stationary phase. Genetics, 2003, 165(4): 2317-2318; author reply 2319-2321.
[53] Sandegren L, Andersson DI. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat Rev Microbiol, 2009, 7(8): 578-588.
[54] Kim N, Jinks-Robertson S. dUTP incorporation into genomic DNA is linked to transcription in yeast. Nature, 2009, 459(7250): 1150-1153.
[55] Gaillard H, Herrera-Moyano E, Aguilera A. Transcriptionassociated genome instability. Chem Rev, 2013, 113(11): 8638-8661.
[56] Datta A, Jinks-Robertson S. Association of increased spontaneous mutation rates with high levels of transcription in yeast. Science, 1995, 268(5217): 1616-1619.
[57] Wright BE, Longacre A, Reimers JM. Hypermutation in derepressed operons of Escherichia coli K12. Proc Natl Acad Sci USA, 1999, 96(9): 5089-5094.
[58] Wright BE. Stress-directed adaptive mutations and evolution. Mol Microbiol, 2004, 52(3): 643-650.
[59] Green P, Ewing B, Miller W, Thomas PJ, Green ED. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet, 2003, 33(4): 514-517.
[60] Diaz M, Lawrence C. An update on the role of translesion synthesis DNA polymerases in Ig hypermutation. Trends Immunol, 2005, 26(4): 215-220.
[61] Cohen SE, Godoy VG, Walker GC. Transcriptional modulator NusA interacts with translesion DNA polymerases in Escherichia coli. J Bacteriol, 2009, 191(2): 665-672.
[62] Cohen SE, Lewis CA, Mooney RA, Kohanski MA, Collins JJ, Landick R, Walker GC. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc Natl Acad Sci USA, 2010, 107(35): 15517-15522.
[63] Cohen SE, Walker GC. The transcription elongation factor NusA is required for stress-induced mutagenesis in Escherichia coli. Curr Biol, 2010, 20(1): 80-85.
[64] McGlynn P. Linking transcription with DNA repair, damage tolerance, and genome duplication. Proc Natl Acad Sci USA, 2010, 107(35): 15314-15315.
(责任编委: 李绍武)
Stress-induced cellular adaptive mutagenesis
Linjiang Zhu, Qi Li
The Key Laboratory of Industrial Biotechnology, Ministry of Education, Synergetic Innovation Center of Food Safety and Nutrition, Jiangnan University, Wuxi 214122, China
The adaptive mutations exist widely in the evolution of cells, such as antibiotic resistance mutations of pathogenic bacteria, adaptive evolution of industrial strains, and cancerization of human somatic cells. However, how these adaptive mutations are generated is still controversial. Based on the mutational analysis models under the nonlethal selection conditions, stress-induced cellular adaptive mutagenesis is proposed as a new evolutionary viewpoint. The hypothetic pathway of stress-induced mutagenesis involves several intracellular physiological responses, including DNA damages caused by accumulation of intracellular toxic chemicals, limitation of DNA MMR (mismatch repair) activity, upregulation of general stress response and activation of SOS response. These responses directly affect the accuracy of DNA replication from a high-fidelity manner to an error-prone one. The state changes of cell physiology significantly increase intracellular mutation rate and recombination activity. In addition, gene transcription under stress condition increases the instability of genome in response to DNA damage, resulting in transcription-associated DNA mutagenesis. In this review, we summarize these two molecular mechanisms of stress-induced mutagenesis and transcription-associated DNA mutagenesis to help bet-ter understand the mechanisms of adaptive mutagenesis.
stress-induced mutagenesis; adaptive evolution; hypermutation; transcription-associated DNA mutagenesis
2013-09-04;
2013-12-23
2013基本科研-青年基金项目(编号:1012050205134030)和中央高校基本科研业务费资金(编号:JUSRP51306A)资助
朱林江, 博士, 副教授, 研究方向:微生物生理及其改造。Tel: 0510-85918176; E-mail: zlj@jiangnan.edu.cn
10.3724/SP.J.1005.2014.0327
时间: 2014-3-27 14:29:48
URL: http://www.cnki.net/kcms/detail/11.1913.R.20140327.1429.001.html
猜你喜欢
农业科技通讯(2023年1期)2023-02-12 07:07:54
遗传(2021年10期)2021-11-01 10:30:08
传染病信息(2021年6期)2021-02-12 01:52:08
中国医学创新(2020年11期)2020-06-08 10:38:37
中华肺部疾病杂志(电子版)(2020年2期)2020-05-07 00:34:24
中国实验诊断学(2020年4期)2020-04-29 14:30:28
中国外汇(2019年23期)2019-05-25 07:06:20
中成药(2017年12期)2018-01-19 02:06:27
福建轻纺(2017年12期)2017-04-10 12:56:39
北京航空航天大学学报(2016年9期)2016-11-16 02:02:33
