
ABS/NBR TPV压缩永久形变的可逆回复机制及模型构建*
2014-05-21 02:17:32魏东亚王兆波
弹性体 2014年6期
魏东亚,赵 静,王兆波
(青岛科技大学 材料科学与工程学院,山东 青岛 266042)
热塑性硫化胶(TPV)是热塑性弹性体(TPE)的一种,它具有常温下的橡胶高弹性和高温下的热塑化性[1-2]。动态硫化法最早由Gessler提出,之后Fisher[3]以及Coran[4]等人分别对部分动态硫化体系和全动态硫化体系进行了深入研究。20世纪80年代Exxon Mobil公司率先实现TPV的商业化生产,并在汽车、建筑、居家用设备、电缆、医疗器械等领域得以应用。但TPV普遍存在的橡胶质感差、永久形变偏大及硬度偏高等缺点影响了其推广。De Risi[5]研究了交联剂含量、橡塑比、压缩温度、增强填料对聚丙烯(PP)/三元乙丙橡胶(EPDM)TPV压缩永久形变的影响,Vennemann[6]发现了提高EPDM交联密度可以显著降低PP/EPDM TPV的压缩永久形变;但到目前为止,关于TPV压缩永久形变可逆性回复的报道却很少。
硫化橡胶在压缩时会发生物理和化学变化。当压缩力去除后,这些变化阻碍橡胶恢复到其初始状态,并由此导致永久形变的产生。压缩永久形变的大小往往取决于压缩状态及回复时的温度与时间。通常在高温条件下,化学变化是导致橡胶发生压缩永久形变的主要原因;而在低温下,玻璃态硬化和结晶形成则是主要影响因素,且当温度回升后,这些作用会消失。对于传统橡胶,压缩永久形变主要取决于其交联密度,但对于TPV,具有“海-岛”型特殊结构,其压缩永久形变不仅与橡胶相的交联密度有关,还取决于基体树脂相的性质,使得TPV压缩永久形变的可逆回复与传统橡胶存在迥然差异。
本课题组前期制备了丙烯腈-丁二烯-苯乙烯三元共聚物(ABS)/丁腈橡胶(NBR)共混型动态硫化体系[7],本实验以其为研究对象,研究了橡塑比和热处理条件对TPV压缩永久形变可逆回复的影响,在此基础上探讨其回复机制,并通过对数据的拟合采用Maxwell模型对压缩永久形变可逆回复过程进行了描述。
1 实验部分
1.1 原料
ABS:牌号EX18T,日本UMG ABS株式会社;NBR:牌号3305,丙烯腈质量分数为35%,中国石油兰州石化公司;其它助剂均为常用工业级配合剂。
1.2 设备与仪器
X(S)K-160型双辊开炼机和50 t平板硫化机:上海群翼橡塑机械有限公司;CH-10型厚度计:扬州市俊平试验机械有限公司;DHG-9023A型鼓风干燥箱:上海和呈仪器制造有限公司。
1.3 试样制备
NBR胶料配方(质量份):NBR 100;S 1.0;TS 1.5;促进剂CZ 1.2;ZnO 5.0;硬脂酸 1.5;防老剂RD 1.0。
(1) 首先在双辊开炼机上将NBR与各种配合剂混炼均匀制成母炼胶,下片。
(2) 将ABS置于165 ℃的开炼机上充分熔融塑化,加入NBR母炼胶,动态硫化5 min,下片。
(3) 动态硫化样品置于模具中在平板硫化机上于180 ℃预热6 min,排气3~5次,保压5 min,冷压8 min后将样品取出。
1.4 压缩永久形变及其可逆回复测试
实验前测量试样初始高度h0(在压缩后样品可逆回复的温度条件下进行测试)。参考GB/T 7759—1996,压缩率为20%,在室温条件下将试样及限制器置于夹具中,均匀压缩至规定高度h1;在室温下放置24 h后,松开夹具紧固件,将试样取下后立刻置于特定温度的烘箱中进行热处理,并定期测试高度h2。瞬时压缩永久形变(K)的计算如式(1)所示:
(1)
2 结果与讨论
2.1 系列动态硫化ABS/NBR体系的压缩永久形变的可逆回复
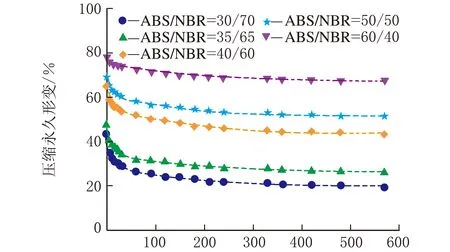
图1是在不同热处理温度下测试的系列ABS/NBR TPV压缩永久形变-时间关系曲线,从曲线变化趋势可以看出压缩永久形变的可逆回复过程。从图1(a)可以看出,压缩后试样在室温条件下,其压缩永久形变在100 min内的可逆回复相对较快,之后则趋于缓慢降低;由图1(a)还可以看出,随着橡塑比的增加,压缩永久形变的可逆性回复显著加快,且压缩永久形变的初始值和最终残余值呈明显下降趋势,这是由于橡胶相的弹性回复能力增强所致。为了促进TPV中发生塑性形变的基体ABS充分解取向,并由此获得更大的形变回复的驱动力,加速压缩形变的可逆回复,特将压缩后的试样置于不同的温度条件下进行热处理。图1(b)~图1(d)分别是在80 ℃、100 ℃与120 ℃的热处理条件下测试的TPV的压缩永久形变-时间关系曲线。由图1(b)~图1(d)可以看出,在热处理条件下的残余压缩永久形变明显减少,表现出明显增强的形变可逆回复;值得注意的是,当热处理温度达到120 ℃时,不同橡塑比的TPV的压缩永久形变均几乎完全可逆。
一般来说,对于处于玻璃态的非晶塑料在室温条件下冷拉产生的塑性形变,在室温条件下是不能回复的,通常需要升温至玻璃化转变温度(Tg)附近形变才能使得可逆回复。需要指出的是,图1(b)与图1(c)的热处理温度虽然远低于ABS的Tg(125 ℃),但压缩永久形变仍能得以快速地较大程度的回复,这表明虽然ABS为TPV的连续相,但是NBR分散相粒子的弹性回复作用加速了压缩永久形变的可逆回复。
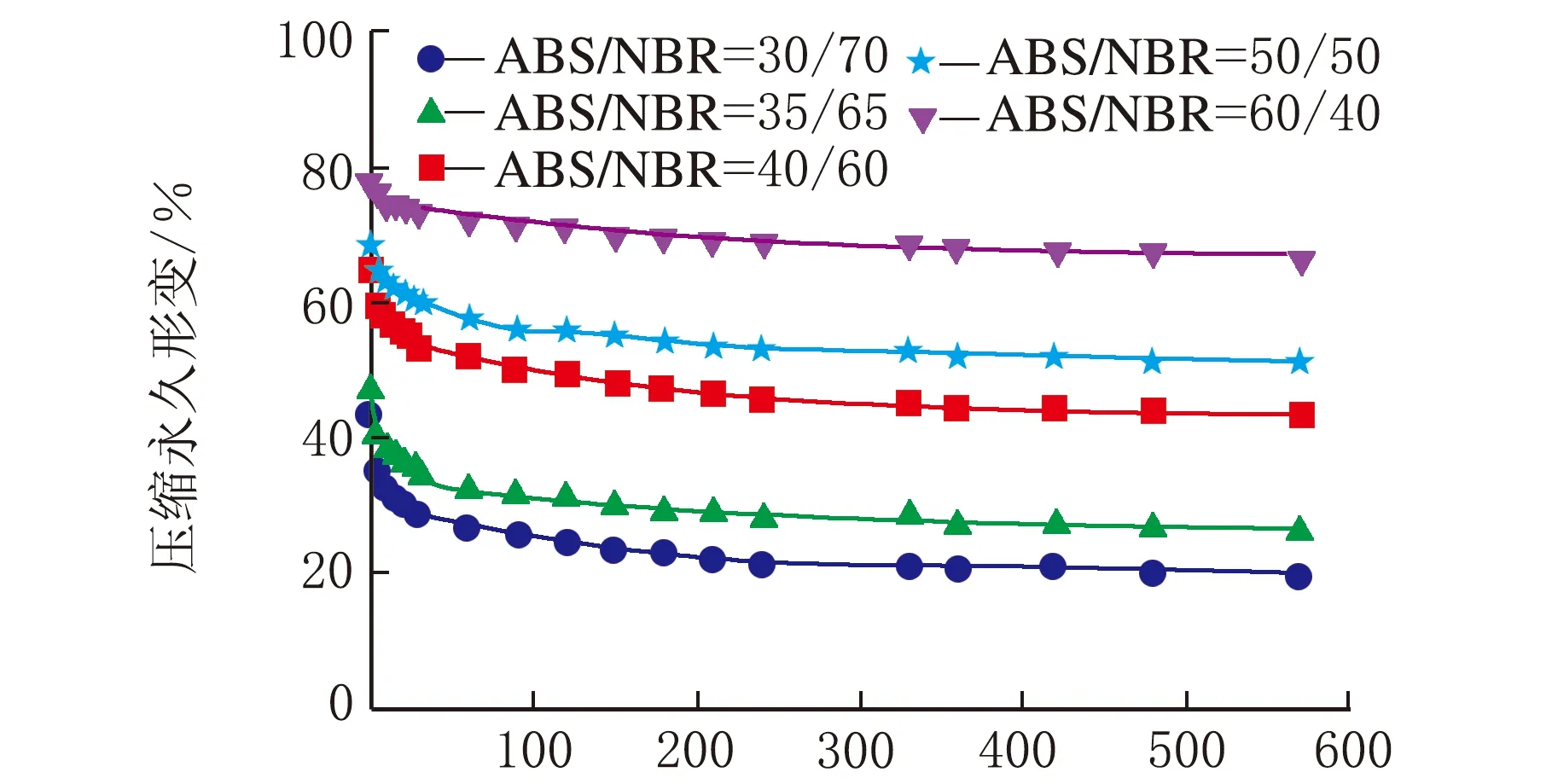
时间/min(a) 23 ℃
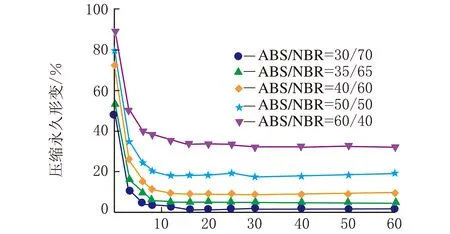
时间/min (b) 80 ℃
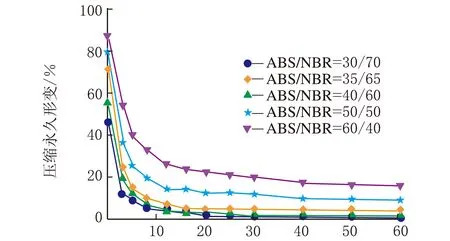
时间/min (c) 100 ℃
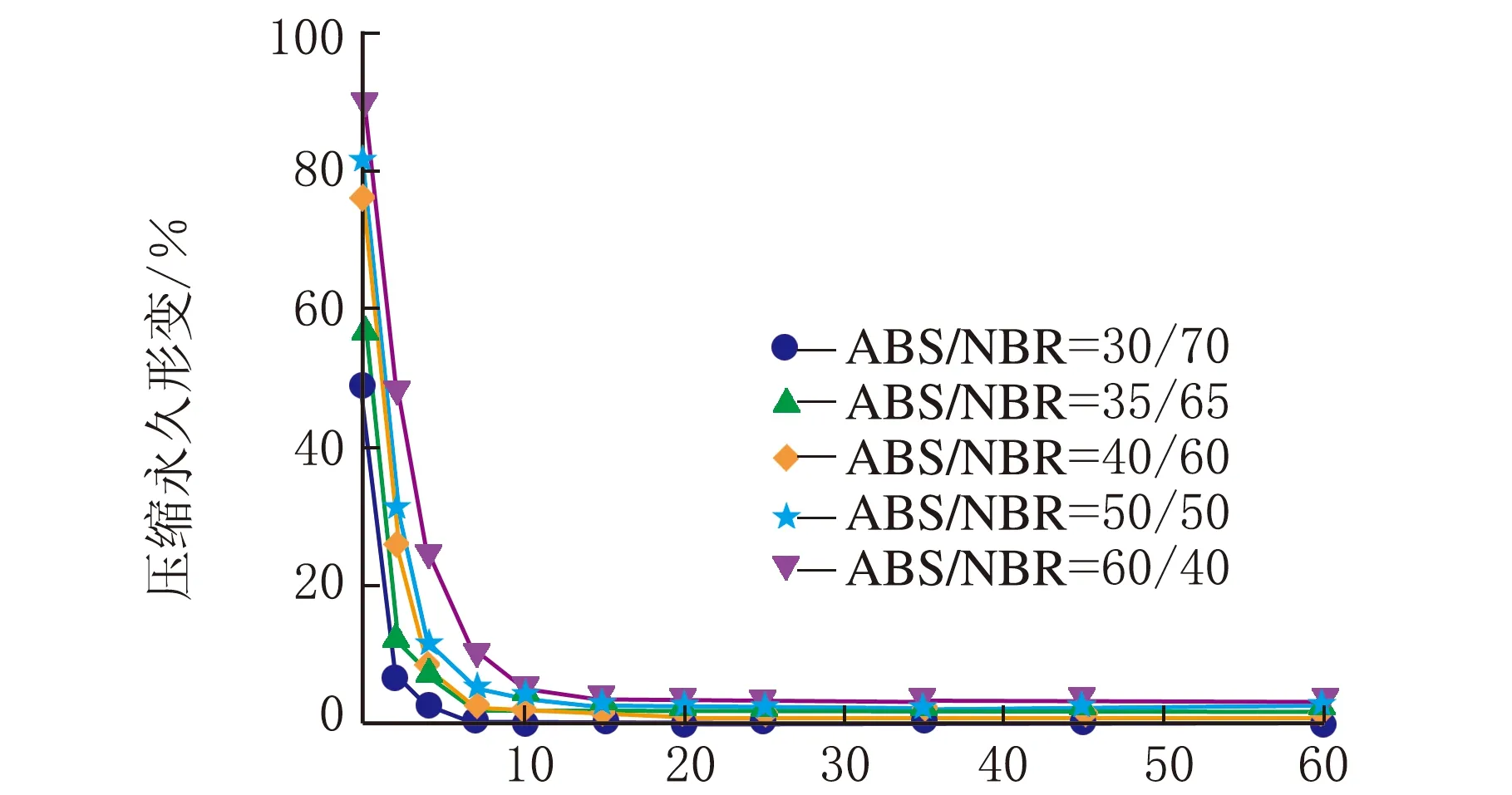
时间/min(d) 120 ℃图1 不同热处理温度下系列ABS/NBR动态硫化体系压缩永久形变-时间关系曲线
2.2 ABS/NBR TPV压缩永久形变可逆回复的机制
从图1中压缩永久形变可逆回复的变化趋势可以看出,TPV压缩永久形变的可逆回复可分为3个阶段:快速回复期、缓慢回复期及平坦期。
为了阐述可逆回复过程,在文献[8]中提出的TPV核-壳模型的基础上,提出图2的TPV压缩永久形变的可逆回复的微观机制示意图,图2中的内部球形粒子代表NBR硫化胶粒子,其外壳则为基体ABS树脂层。
高分子材料在一定的条件下处于一定的分子运动状态,改变条件就能改变分子运动状态,这种分子运动状态的改变在动力学上称为松弛,伴随着松弛,高分子的物理性质发生了急剧转变。当对TPV进行压缩时,其树脂相和橡胶相中的分子构象均发生相应转变。对于基体ABS,在室温压缩时,由于压缩时温度远低于其Tg,ABS中分子运动的内摩擦力很大,压缩时导致分子发生取向,在外力去除后,难以自发发生解取向。另一方面,在压缩过程中,ABS树脂层所发生的取向程度也并不一致,图2中橡胶粒子上下面的树脂相[如图2(b)阴影部分]由于受力最大,产生的塑性形变也因而较大,取向程度较高[9-10]。对于硫化胶粒子,在TPV受到压缩时,树脂相发生形变时也将压力通过界面传递给橡胶相,从而带动其随之发生形变,且形变较大[11]。由于橡胶相为交联结构,分子链间不能滑移,所以应力不会松弛到零,只能松弛到某一数值。
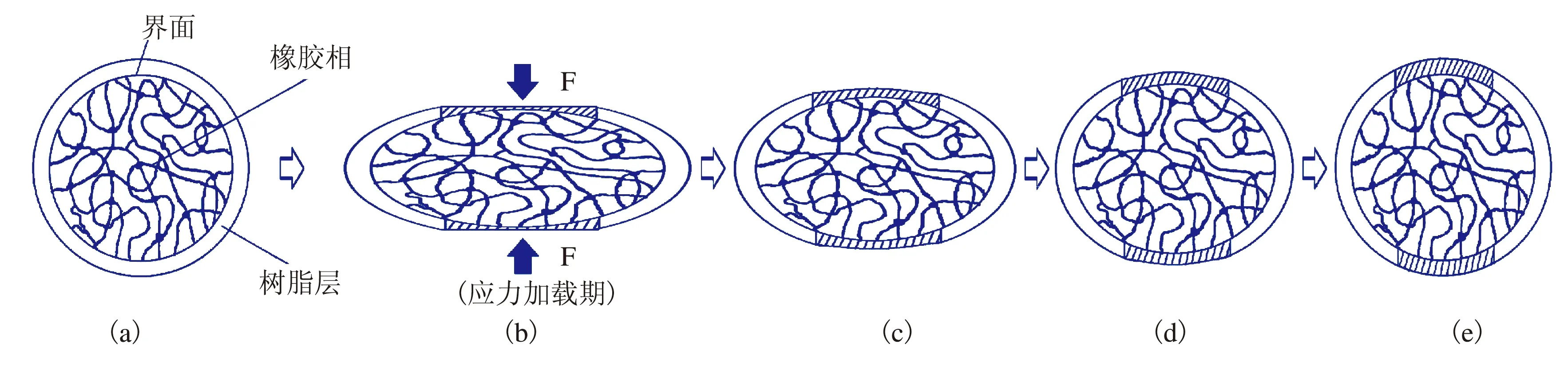
图2 TPV压缩永久形变可逆回复的微观机制模型
当压缩后试样从夹具中取出即处于自由状态,由于橡胶相在压缩过程时发生较大形变,从而产生强的回弹力,且在橡胶相发生弹性回复的同时,通过界面作用带动发生塑性形变的树脂相也产生一定的回复。图2(b)中阴影部分树脂相的塑性形变最大,受到橡胶粒子传递的回复拉动作用也强,因而这部分形变的回复较快,这应该是可逆回复的第一个阶段最主要的回复,如图2(b)和图2(c)所示。在缓慢回复阶段,如图2(c)~图2(d)所示,由于橡胶相在弹性回复的过程中,回弹力随着橡胶相形变的减少而逐渐减弱,另外图2(c)中阴影部分的取向度也会随着压缩永久形变的回复而降低,导致形变回复减缓。在形变回复的第三阶段,橡胶相的形变大部分已得到回复,此时压缩永久形变的回复主要是图2(d)中非阴影部分的树脂相的解取向而导致的回复(这种方式的解取向贯穿于压缩永久形变回复的全过程,但在形变可逆回复的前二阶段对回复贡献较小),如图2(d)和图2(e)所示。
在同一温度下,树脂相较多时TPV的压缩永久形变回复较慢且残余形变较大,这主要是因为当树脂相含量较高时,发生解取向与塑性形变的回复所需要外力较高,加之橡胶相含量较低,产生的弹性回复力也较弱,导致压缩永久形变的回复减慢且残余形变较大。
对于同一样品,压缩后的热处理温度越高,压缩永久形变回复越快且残余压缩永久形变越小。当热处理温度较高时,橡胶相的弹性模量增加,此时橡胶相产生的回弹力增强,传递给树脂相的回复力也相应增加;其次,由于温度升高基体树脂相的分子间的运动更容易,解取向容易发生,形变可逆回复的驱动力增大,这两者同时作用,加速了压缩永久形变的可逆回复。
2.3 ABS/NBR TPV压缩永久形变可逆回复的模型拟合
广义Maxwell模型[12]是描述高分子材料粘弹行为常用的数学模型,一般是用来说明应力、模量或刚度随时间的变化规律,结合2.2节中对TPV压缩永久形变的可逆回复机制的探讨,广义Maxwell模型对TPV压缩永久形变可逆回复过程的描述,如公式(2)所示:
K(t)=k1e-t/τ1+k2e-t/τ2+k3e-t/τ3+k0
(2)
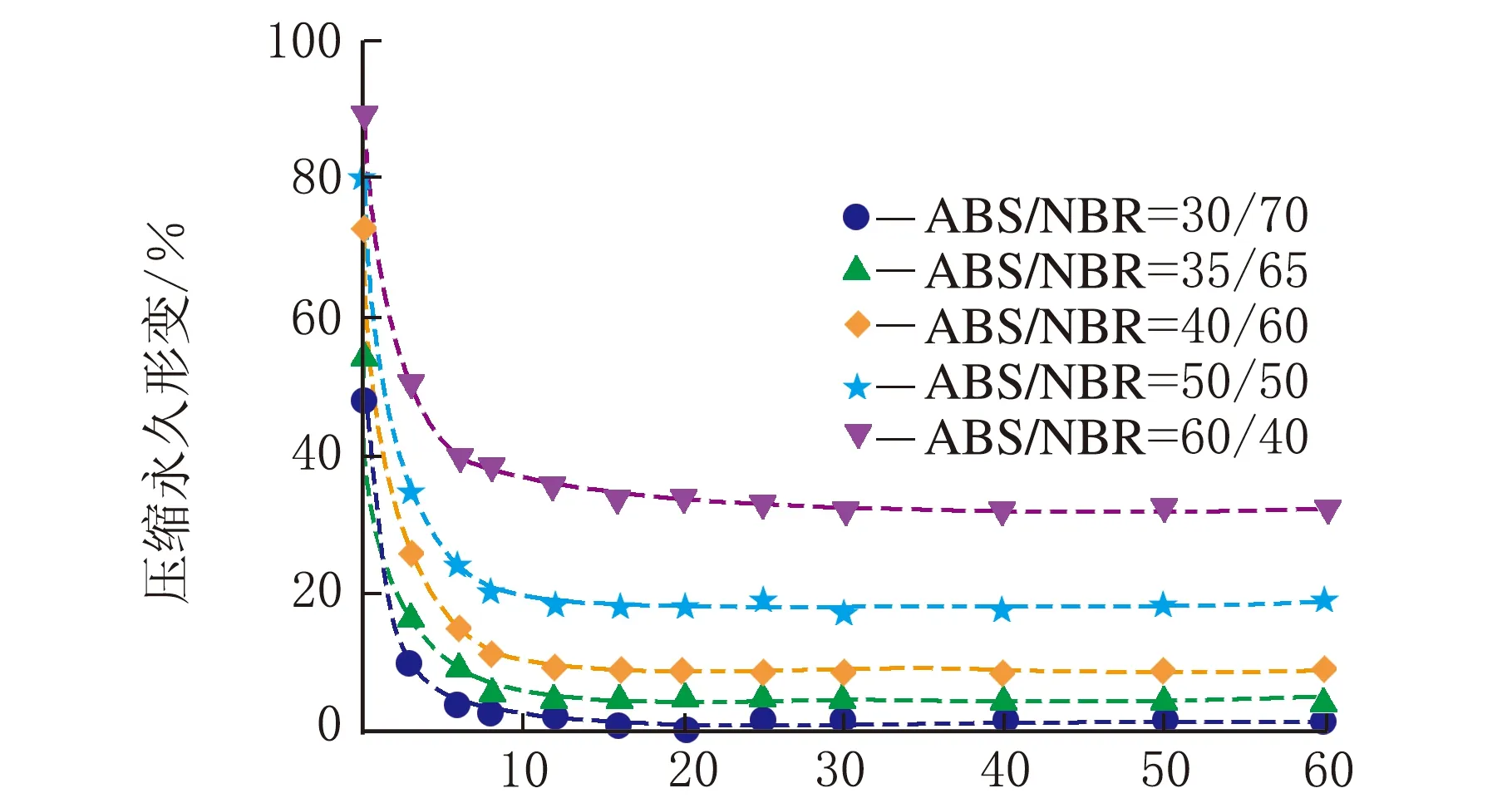
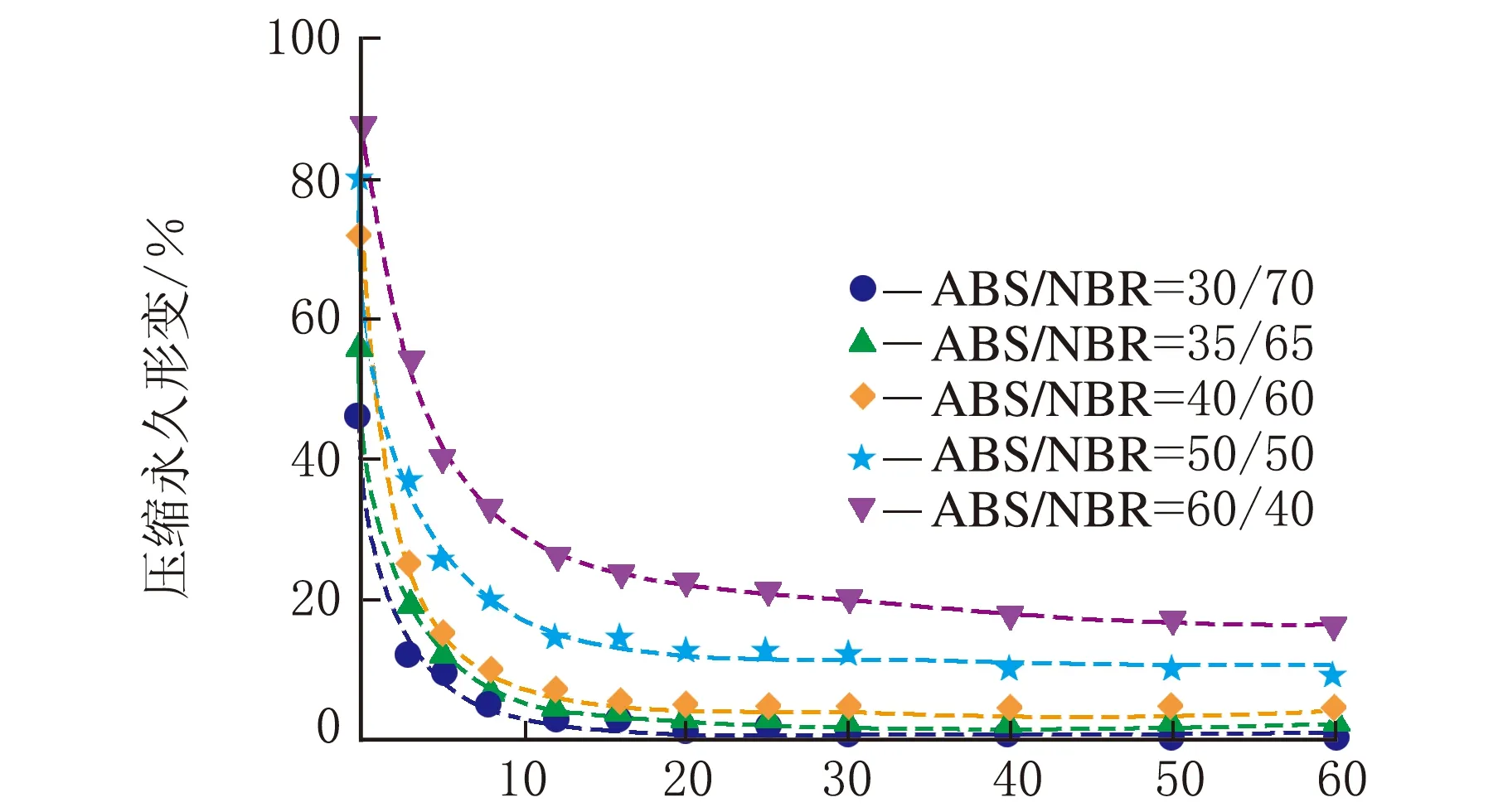
根据公式(2),采用Origin 8.0软件对图1中数据进行拟合处理。结合上节中构建的TPV压缩永久形变可逆回复物理模型,公式(2)中的各参数的物理含义如下:K表示瞬时压缩永久形变,τ1、τ2和τ3分别为可逆回复3个阶段的松弛时间,k1、k2与k3分别表示可逆回复中各段所占比例,k0为实验条件下的压缩永久形变的不可逆部分。公式(2)对图1中的测试数据进行拟合,所得结果如图3所示,其中图符为实测数据点,而虚线则为采用公式(2)拟合出的曲线;由图3可以看出,该数学模型可以很好地描述压缩永久形变的可逆回复过程。拟合过程所获得的各参数的数值见表1~表4。
时间/min(a) 23 ℃
时间/min (b) 80 ℃
时间/min (c) 100 ℃
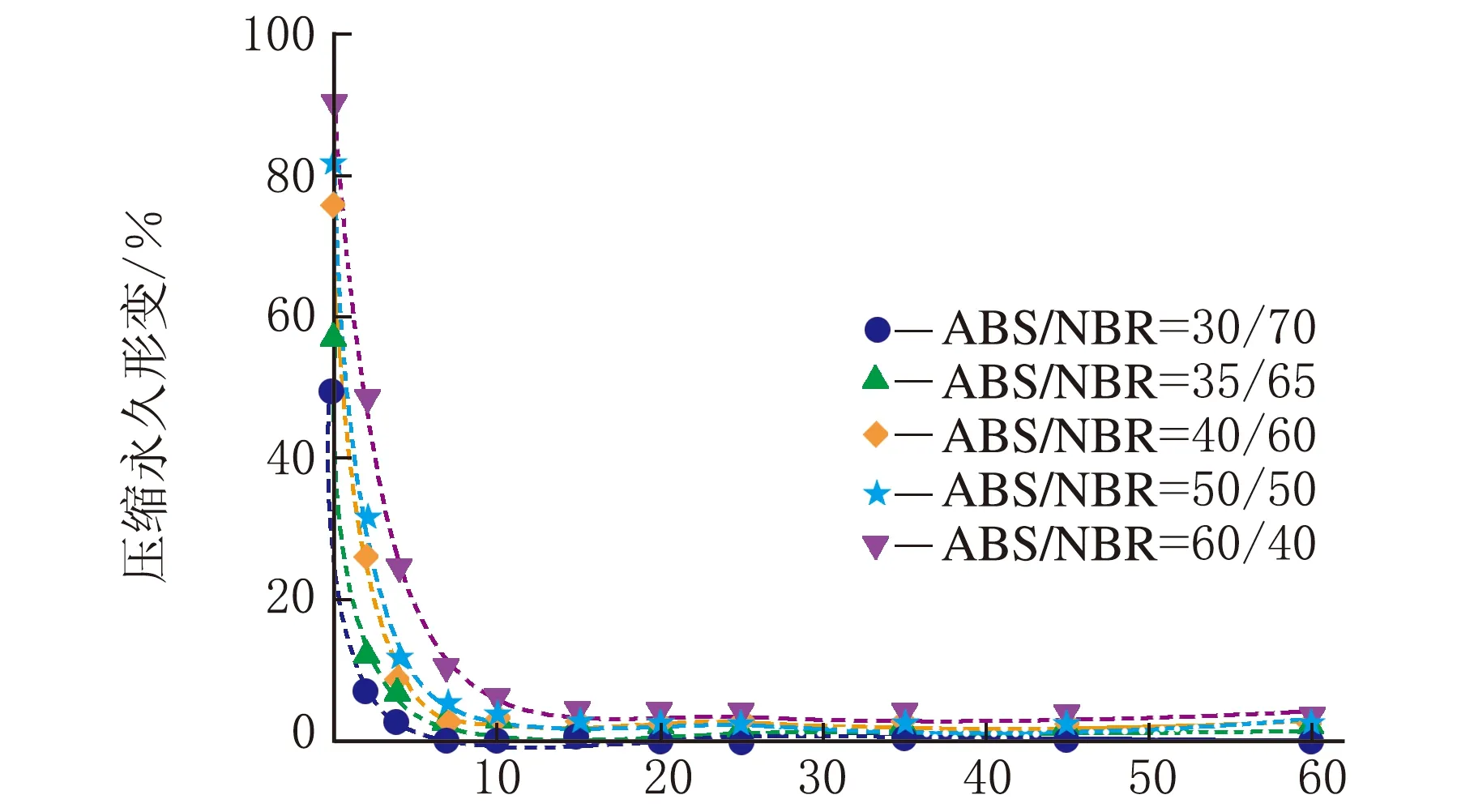
时间/min(d) 120 ℃图3 ABS/NBR动态硫化体系的压缩永久形变回复的拟合曲线
从表1~表4可以看出,对于同一体系,其压缩永久形变回复的τ1、τ2和τ3依次增加,表明压缩永久形变的3个阶段的回复所需时间依次增多,这与图2中模型是一致的。随着橡塑比的降低,k0增大;但是提高热处理温度,k0则发生大幅度降低,表明不可逆部分减少。对于不同橡塑比及不同热处理条件下的样品的τ1、τ2和τ3值,数据波动,未发现明显的规律性。这可能是由于形变回复过程中的复杂因素所致,在室温条件下,取向的基体难以发生解取向,并成为TPV形变可逆回复的阻碍,可逆回复的动力主要来源于橡胶相的弹性回复作用;但在升温尤其靠近基体Tg的条件下,经压缩取向的基体在热处理条件下却容易发生解取向,并为TPV形变回复提供了新的驱动力,而且温度的提高,也使得橡胶相的回复力增强与TPV两相之间界面强化,最终使得体系的可逆回复部分明显提高。对于不同橡塑比的TPV,其橡胶粒子尺寸和树脂层厚度存在差异,且在不同热处理条件下进行测试,基体的解取向容易程度、可逆回复各段基体的解取向度、TPV的界面作用以及橡胶相的弹性回复力也存在差异,这些复杂的因素共同影响了可逆回复各阶段所占比例及松弛时间,并导致规律性不显著。
总体而言,提高热处理温度可大大加速压缩永久形变的可逆回复,且在Tg附近时压缩永久形变可实现完全可逆;该数学模型可以较好地描述压缩永久形变可逆回复的各个阶段所占比例及各段松弛时间,这为压缩永久形变的可逆回复的深入研究提供依据。
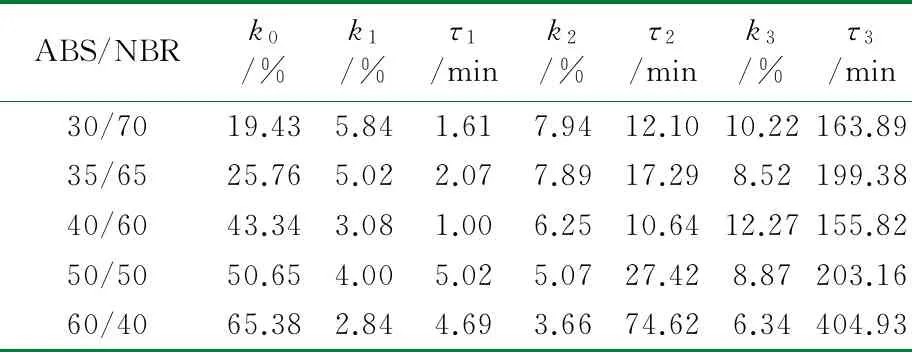
表1 室温下ABS/NBR动态硫化体系压缩永久形变回复拟合数据
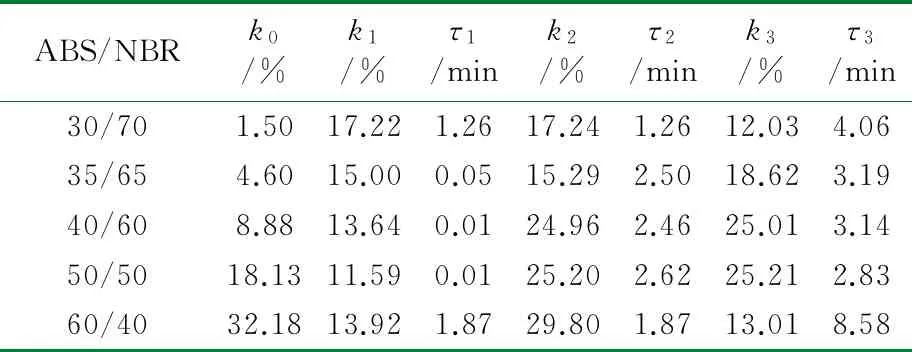
表2 80 ℃下ABS/NBR动态硫化体系压缩永久形变回复拟合数据
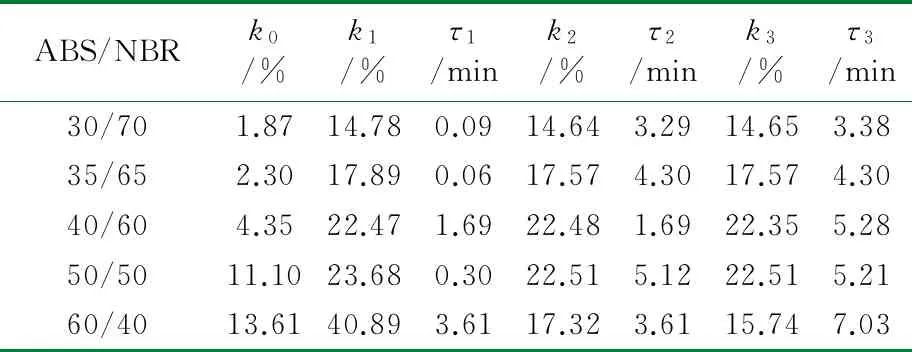
表3 100 ℃下ABS/NBR动态硫化体系压缩永久形变回复拟合数据
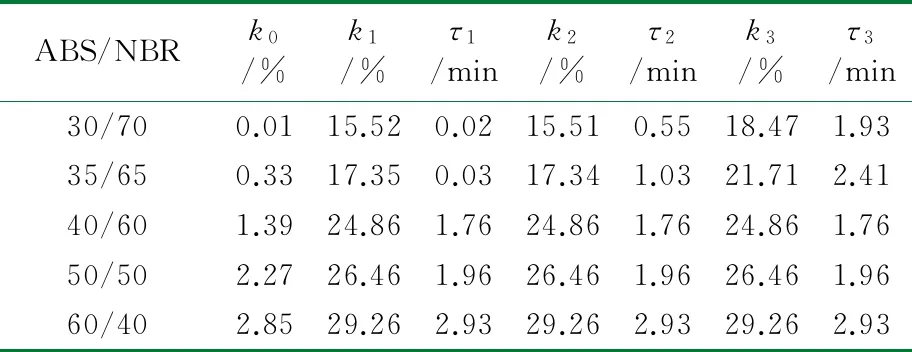
表4 120 ℃下ABS/NBR动态硫化体系压缩永久形变回复拟合数据
3 结 论
(1) 提高橡塑比和热处理温度,ABS/NBR TPV压缩永久形变的可逆回复明显加快,且残余形变较小;在热处理条件下,高橡塑比TPV的压缩永久形变可实现完全可逆回复。
(2) 首次提出TPV压缩永久形变可逆回复的物理模型,橡胶相的弹性回复和树脂层的解取向,是TPV形变回复的驱动力。
(3) 在对实验数据拟合的基础上,采用Maxwell模型对压缩永久形变可逆回复过程进行了描述,并对可逆回复的3个阶段的松弛时间、可逆回复的比例和不可逆部分进行了定量的表征。
参 考 文 献:
[1] 程相坤,赵洪玲,王兆波.EPDM/HDPE热塑性硫化胶的结构与性能研究[J].弹性体,2010,20(1):65-69.
[2] 王利杰,郎丰正,杜芳林,等.低硬度 CM/EVA TPV 的制备及性能[J].弹性体,2013,22(6):47-51.
[3] Fischer W K.Thermoplastic blend of partially cured monoolefin copolymer rubber and polyolefin plastic:US,3758643[P].1973-09-11.
[4] Coran A Y,Patel R P,Williams D.Rubber-Thermoplastic compositions.Part V.Selecting polymer for thermoplastic vulcanizates[J].Rubber Chemistry and Technology Journal,1982,55(1):116-136.
[5] De Risi F R,Noordermeer J W M.Effect of methacrylate co-agents on peroxide cured PP/EPDM thermoplastic vulcanizates[J].Rubber Chemistry and Technology,2007,80(1):83-99.
[6] Vennemann N,Bökamp K,Bröker D.Crosslink density of peroxide cured TPV[J].Macromolecular Symposia,2006,245(1):641-650.
[7] Wei Dongya,Mao Changming,Li Shuai,et al.Dynamically vulcanized nitrile butadiene rubber/acrylonitrile-butadiene-styrene terpolymer blends compatibilized by styrene-butadiene-styrene block copolymer[J].Journal of Macromolecular Science:Part B,2014,53(4):601-614.
[8] Duin M V.Recent developments for EPDM-based thermoplastic vulcanizates[J].Macromolecular Symposia,2006,233(1):11-16.
[9] Oderkerk J,Groeninckx G,Soliman M.Investigation of the deformation and recovery behavior of nylon-6/rubber thermoplastic vulcanizates on the molecular level by infrared-strain recovery measurements[J].Macromolecules,2002,35(10):3946-3954.
[10] Oderkerk J,de Schaetzen G,Goderis B,et al.Micromechanical deformation and recovery processes of nylon-6/rubber thermoplastic vulcanizates as studied by atomic force microscopy and transmission electron microscopy[J].Macromolecules,2002,35(17):6623-6629.
[11] Huy T A,Luepke T,Radusch H J.Characterization of the deformation behavior of dynamic vulcanizates by FTIR spectroscopy[J].Journal of Applied Polymer Science,2001,80(2):148-158.
[12] Siengchin S,Karger-Kocsis J.Mechanical and stress relaxation behavior of SantopreneRthermoplastic elastomer/boehmite alumina nanocomposites produced by water-mediated and direct melt compounding[J].Composites:Part A,2010,41(6):768-773.
猜你喜欢
石材(2022年3期)2022-06-01 06:23:54
原道(2022年2期)2022-02-17 00:59:12
军民两用技术与产品(2021年8期)2021-11-24 01:09:34
理化检验-化学分册(2020年5期)2020-06-15 11:36:04
模具制造(2019年10期)2020-01-06 09:13:08
中国特种设备安全(2019年3期)2019-04-22 05:05:38
电镀与环保(2018年4期)2018-08-20 03:08:02
山东工业技术(2016年15期)2016-12-01 05:30:43
焊接(2016年2期)2016-02-27 13:01:20
山东冶金(2015年5期)2015-12-10 03:27:41
