
氯取代基对经典Diels-Alder反应影响的密度泛函研究
2014-05-09 09:33姚树文崔乘幸张艺川
河南科技学院学报(自然科学版) 2014年6期
姚树文,崔乘幸,张艺川
(1.河南科技学院,河南新乡453003;2.河南师范大学化学化工学院,河南新乡453000)
1928年,狄尔斯与阿尔德在研究顺丁烯二酸酐与1,3-丁二烯反应时,发现三键或双键化合物在与共轭双烯相互作用时会生成六元环状化合物,这类反应即Diels-Alder(DA)反应,也称为双烯合成反应[1].无取代的DA反应中共轭双烯为供电子体,不饱和键为受电子体.DA反应在温和条件下就可以得到六元环,是有机化学合成反应中非常重要的碳碳键形成的手段之一,也是现代有机合成里常用的反应之一[2-6].该反应有丰富的立体化学呈现,兼有立体选择性、立体专一性和区域选择性等.关于DA反应的机理直到现在还没有一致的结论.可能存在的反应机理有协同反应机理、分步反应机理、协同和分步反应机理共存3种.
顺式-1,3-丁二烯和乙烯之间的反应是经典的Diels-Alder反应,很少有人对反式-1,3-丁二烯和乙烯之间的反应的研究发表结论[7].本文同时研究了顺式-1,3-丁二烯与反式-1,3-丁二烯与氯代乙烯的Diels-Alder反应.基于经典的Diels-Alder反应,通过比较反应中各个稳定点的构型和能量,研究氯取代基对经典的Diels-Alder反应势能面的影响,分析该反应的过程和机理.
1 计算方法
经典Diels-Alder反应见图1.

图1 经典的Diels-Alder反应Fig.1 The iconic Diels-Alder reaction
图1中R代表不同的取代基,吸电子基团和斥电子基团对反应过程有不同的影响.这里考虑了吸电子基团氯取代乙烯时,反应机理所受到的影响.在M06-2X/6-31+G(d)理论水平下对3种氯代乙烯与1,3-丁二烯的Diels-Alder反应进行计算.反应中协同反应路径使用闭壳层,采用限制性计算方法.分步双自由基反应路径使用开壳层,采用非限制性方法结合对称性破损技术进行计算.所有的计算都在Gaussian 03中进行.
2 结果与讨论
2.1 顺式-1,3-丁二烯的反应
图2所示是1,3-丁二烯与氯代乙烯反应的完整过程图.

图2 顺式-及反式-1,3-丁二烯和氯代乙烯反应Fig.2 The DA reaction of cis-and trans-1,3-butadiene and ethylene
图2中反式-1,3-丁二烯与氯代乙烯的反应在上方,顺式-1,3-丁二烯与氯代乙烯的反应在下方反应过程中各个稳定点的名称用 cis、trans、R、int、TS、P、bi和 con 八个关键词定义.关键词 R、P、bi和 con分别表示反应物、产物、协同和双自由基分步.例如:cis-bi-TS代表顺式结构的1,3-丁二烯(trans-R)与氯代乙烯反应的第一个TS,并且这个TS在协同反应中.TS的结构和名称标在箭头的上面,反应物、过渡态、产物的结构和名称标在箭头之间.所有的稳定点,分别以反式1,3-丁二烯和乙烯之和,反式1,3-丁二烯和氯代乙烯能量之和,反式1,3-丁二烯和1,1-二氯乙烯之和,反式1,3-丁二烯和反式-1,2-二氯乙烯反应物的相对能量都用吉布斯自由能表示(ΔG),列于表1中.

表1 在M06-2X/6-31+G(d)理论水平下反应坐标上各稳定点的相对能量Tab.1 The relative Gibbs free energy of each stationary points on the potential energy curve for the four reactions kcal/mol
先研究讨论1,3-丁二烯与氯代乙烯,1,1-二氯乙烯,反-1,2-二氯乙烯的反应过程,即图2下半部分.经典的1,3-丁二烯与乙烯反应有2种可能的反应过程,即协同反应过程和双自由基分步反应过程.对于协同反应,顺式1,3-丁二烯与氯代乙烯的取代反应可参见图2中下半部分的反应过程.氯代乙烯与顺式1,3-丁二烯的DA反应的主要稳定点的构型如图3所示.

图3 顺式1,3-丁二烯与氯代乙烯反应势能曲线上主要稳定点的结构参数Fig.3 The dominant geometry parameters of stationary points on the potential energy surface of cis-1,3-butadiene and chloride ethylene
从图3中的结构变化可以发现,在协同反应路径中,只优化得到一个过渡态cis-con-TS.由于Cl取代基的影响,cis-con-TS中2个新形成碳碳单键键长分别为0.222 3 nm和0.228 1 nm,不再等长.此反应为协同非同步反应.顺式-1,3-丁二烯与1,1-二氯乙烯、反式-1,2-二氯乙烯在协同反应路径中,同样只优化得到一个过渡态cis-con-TS.受取代基影响,1,1-二氯代乙烯反应中cis-con-TS两个新形成的碳碳键键长分别为0.220 9 nm和0.232 9 nm,而反1,2-二氯代乙烯反应中cis-con-TS两个新形成碳碳键键长分别为0.222 7 nm和0.227 2 nm.在M06-2X/6-31+G(d)理论水平下对顺式-1,3-丁二烯与氯代乙烯,1,1-二氯乙烯,和反式-1,2-二氯乙烯反应过程中协同反应的能垒进行计算分别为31.6、32.6和33.1 kcal/mol.
分步反应过程中cis-R经过cis-bi-TS1生成cis-bi-int,再经过cis-bi-TS2生成cis-P.对氯乙烯的反应,cis-bi-TS1中新形成的碳碳键键长为0.190 0 nm,cis-bi-int中为0.152 5 nm.另一个碳碳单键没有形成.cis-bi-int经过cis-bi-TS2生成cis-P.cis-bi-TS2新生成的碳碳单键键长为0.155 4 nm.经过IRC计算证明,cis-bi-TS2是cis-bi-Int与cis-P中间产物.在1,1-二氯乙烯,和反式-1,2-二氯乙烯的反应过程中,cis-bi-TS1中新形成的碳碳单键键长分别为0.1927nm和0.1863nm,cis-bi-int中分别为0.1526 nm和0.1555nm.另一个碳碳单键没有形成.cis-bi-TS2新生成的碳碳键键长分别为0.156 7 nm和0.155 4 nm.顺式-1,3丁二烯与氯代乙烯,1,1-二氯乙烯,和反式-1,2-二氯乙烯反应过程中分步反应的能垒进行计算.cis-R到cis-bi-int所经历cis-bi-TS1三个反应的能垒分别为:45.0、45.4和41.8 kcal/mol.cis-bi-int到cis-P所经历cis-bi-TS2的反应能垒分别为41.0、42.2和37.1 kcal/mol.
2.2 反式-1,3丁二烯的反应
氯代乙烯与反式-1,3-丁二烯的反应势能曲线上主要稳定点的构型见图4.
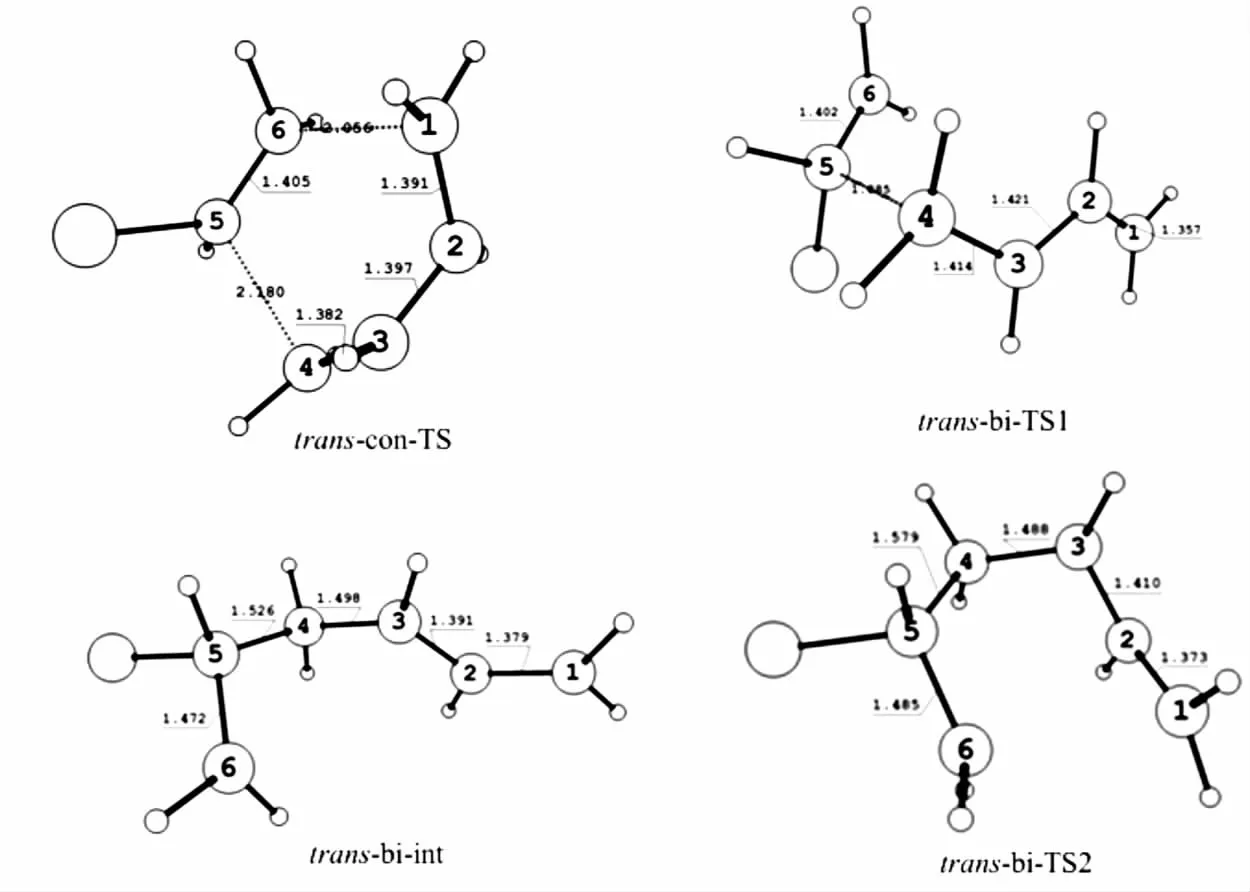
图4 反式-1,3-丁二烯与氯代乙烯反应势能曲线上主要稳定点的结构参数Fig.4 The dominant geometry parameters of stationary points on the potential energy surface of trans-1,3-butadiene and chloride ethylene
从图4中的结构变化可以发现,由于取代基的存在,trans-con-TS中新形成的碳碳键C1-C6的键长是0.205 5 nm,C4-C5的键长为0.223 1 nm,反应过程中π键的键能同样也在减少,σ键的键能在增加.由于反式-1,3-丁二烯的扭转及反应的进行,C2-C3之间由sp2杂化转变为sp3杂化.由于共轭效应的影响,trans-R中间C2-C3之间的碳碳单键键长为0.146 2 nm,比单纯的碳碳单键(0.153 0~0.155 0 nm)略短.而在trans-con-TS中C2-C3之间的键长为0.139 8 nm,更接近碳碳双键的键长.分步反应过程中trans-R经过trans-bi-TS1生成trans-bi-int,再经过trans-bi-TS2生成trans-P.新形成的碳碳键在trans-bi-TS1中为0.187 6 nm.trans-bi-Int新形成的碳碳键长度为0.152 6 nm,而另一个碳碳键此时还没有形成.trans-bi-TS2中即将要形成的2个碳碳键键长分别为0.157 9 nm和0.248 2 nm,然后生成产物trans-P.
分步反应的能垒进行计算.对氯乙烯的反应,trans-R到trans-bi-int所经历trans-bi-TS1为44.5kcal/mol,trans-bi-int到trans-P所经历trans-bi-TS2的反应能垒为56.4 kcal/mol.对二氯乙烯的反应,trans-R到trans-bi-int所经历 trans-bi-TS1a和 trans-bi-TS1b的能垒分别为 44.8 kcal/mol和 45.4 kcal/mol,trans-bi-int到trans-P所经历trans-bi-TS2的反应能垒为56.0 kcal/mol.在反式二氯乙烯的反应中trans-R到trans-bi-int所经历trans-bi-TS1为42.3 kcal/mol,trans-bi-int到trans-P所经历trans-bi-TS2的反应能垒为51.9 kcal/mol.
2.3 两反应之间的转换
顺式-1,3-丁二烯与反式-1,3-丁二烯的分步反应中所对应的中间过渡态trans-bi-int和cis-bi-int之间的转换见图1,中间部分所示,int-bi-TS即为转换的中间驻点,转换过程主要是1,3-丁二烯中2号、3号碳原子之间碳碳键的扭转而实现3个取代反应中trans-bi-int与cis-bi-int之间的转换分别需要经过15.6、15.0和15.1 kcal/mol的能垒.trans-P与cis-P之间的转换分别需要分别经过13.0、9.1和10.3 kcal/mol的能垒.
3 小结
本文研究了氯取代基对DA反应机理的影响,经过研究发现氯取代基由于较强的电负性,对反应中势能面上各个稳定点的构型及能量都有影响,但是并没有改变反应的性质,所有3个反应顺式反应能量上可行,并且以协同的方式进行.
[1]Diels O,Alder K.Synthesen in der hydroaromatischen reihe[J].Justus Liebigs Annalen der Chemie,1928,460:98-122.
[2]Jiang H,Cruz D C,Li Y,et al.Asymmetric organocatalytic Thio-Diels-Alder reactions via trienamine catalysis[J].Journal of the American Chemical Society,2013,135:5200-5207.
[3]Jiang X,Wang R.Recent developments in catalytic asymmetric Inverse-Electron-Demand Diels-Alder reaction[J].Chemical Reviews,2013,113:5515-5546.
[4]Peng F,Grote R E,Wilson R M,et al.Pattern recognition analysis in complex molecule synthesis and the preparation of iso-Diels-Alder motifs[J].Proceedings of the National Academy of Sciences of the United States of America,2013,110:10904-10909.
[5]Qi C,Cong H,Cahill K J,et al.Biomimetic dehydrogenative Diels-Alder cycloadditions:Total syntheses of brosimones A and B[J].Angewandte Chemie International Edition,2013,52:8345-8348.
[6]Sato S,Maeda Y,Guo J D,et al.Mechanistic study of the Diels-Alder reaction of paramagnetic endohedral metallof ullerene:Reaction of La@C82 with 1,2,3,4,5-Pentamethylcyclopentadiene[J].Journal of the American Chemical Society,2013,135:5582-5587.
[7]Bradley A Z,Kociolek M G,Johnson R P.Conformational selectivity in the Diels-Alder cycloaddition:Predictions for reactions of s-trans-1,3-butadiene[J].Journal of Organic Chemistry,2000,65:7134-7138.
猜你喜欢
中国石油石化(2022年10期)2022-06-21
中国特种设备安全(2022年1期)2022-04-26
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
今日农业(2019年11期)2019-08-13
价值工程(2017年31期)2018-01-17
中国司法鉴定(2017年5期)2017-10-11
化工管理(2017年18期)2017-03-03
化工设计通讯(2017年9期)2017-03-02
