
超高效亲水作用色谱-串联质谱法检测水中的苦味酸及苦氨酸
2014-05-08 11:14:56钱飞中朱丽波徐能斌冯加永洪正昉徐立红陈钟佺汪晟乐
色谱 2014年5期
钱飞中, 朱丽波, 徐能斌, 冯加永*, 洪正昉,徐立红, 陈钟佺, 汪晟乐
(1.宁波市环境监测中心,浙江宁波315012;2.浙江省环境监测中心,浙江杭州310012;3.绍兴市环境监测中心,浙江绍兴312000)
2,4,6-三硝基苯酚又名苦味酸,是早期炸药的主要成分,第二次世界大战期间生产的苦味酸炸弹近些年仍然在德国、中国等地时有发现[1]。同时苦味酸广泛用作染料、医疗中的收敛剂和杀菌剂等,也是许多芳香族硝基化合物生产过程中的副产物[2]。苦味酸具有强氧化性和强酸性,在环境中很快降解成毒性更大的苦氨酸等降解产物[3]。
苦味酸是我国地表水常规监测化合物,目前的国标方法采用衍生化-气相色谱法检测水体中的苦味酸[4]。该方法首先需要将沸点较高(300℃)的苦味酸衍生成沸点较低(112℃)的氯化苦,采用气相色谱-电子捕获检测器(ECD)进行检测。该方法存在3个比较大的缺陷,一是需要用毒性较大的苯进行萃取;二是用于反应的次氯酸钠不稳定,衍生反应难以控制,且氯化苦是一种极易爆炸的物质,具有一定的危险性;三是采用 ECD难以对目标物准确定性。
由于苦味酸是一种强酸和强极性化合物,其在25℃时电离常数达0.42,在水中能够较为充分的电离,以至于在普通反相色谱柱中难以实现有效保留[5]。为了尽可能地使其在反相色谱柱中保留,需要在初始流动相中使用很高比例的水相,以降低洗脱能力,使得方法的灵敏度较低[6]。在以往的文献报道中对于苦味酸的保留通常有两种方式[7]:一种采取离子抑制的方法,即在流动相中添加酸性缓冲盐以抑制苦味酸的电离,如Nipper等[8]使用组成为35%(v/v)甲醇和65%(v/v)醋酸钠(pH为4.8)的流动相分析苦味酸;另一种方法是使用离子对试剂,通过离子对试剂与待测离子结合增加其疏水性,从而增加待测离子在反相色谱柱中的保留,如Yost等[9]使用组成为50%(v/v)乙腈和50%(v/v)0.05 mol/L辛烷磺酰的流动相,且流动相中含有0.1 mol/L的三丁基氢氧化铵(tributyl ammonium hydroxide,TBAH)离子对试剂。通常使用的离子对试剂TBAH并不适合液相色谱-质谱(LC-MS)分析,因为不挥发的离子对试剂会在LC-MS接口处沉淀而污染离子源。所以在Pamme等[7]的实验中使用了三丁基甲酸铵(tributyl ammonium formate,TBAF)这种挥发性的离子对试剂。但是,离子对试剂的使用一方面需要考虑其是否能够完全反应,另一方面如果离子对在进样口处不能及时完全挥发仍然可能导致仪器的污染。以上所述的液相方法及衍生化LC-MS方法中,由于仪器灵敏度只能达到mg/L 级[7-9],因此必须采取高倍浓缩、净化等方式进行样品前处理,需要时间较长,过程复杂。
亲水作用色谱主要针对在普通反相色谱中保留弱或者不保留的极性物质,并且可以缓解在反相液相色谱中流动相中高水相引起的反压高及质谱信号低等问题[10]。本文建立了超高效亲水作用色谱-串联质谱法检测水中苦味酸及其降解产物苦氨酸的方法,实现了待测物的有效保留。本方法无需衍生,选择性好,灵敏度高,分析速度快。
1 实验部分
1.1 仪器与试剂
Acquity UPLC超高效液相色谱系统,Quattro Premier XE三重四极杆质谱仪(Waters公司);VisiprepTMSPE真空固相萃取装置(Supelco公司);Oasis MAX固相萃取小柱(200 mg/6 mL,Waters公司);BEH HILIC色谱柱(100 mm×2.1 mm,1.7 μm,Waters公司)。1.0 mL一次性无菌注射器(浙江玉升医疗器械有限公司);有机相针式滤器(13 mm×0.2 μm,上海安谱科学仪器有限公司)。
苦氨酸、苦味酸(99.5%,AccuStandard公司);甲醇、乙腈、甲酸(HPLC级,TEDIA公司);醋酸、氨水(优级纯,国药集团);实验用水为去离子水。
1.2 样品采集及前处理
地表水样品12个,采自浙江省宁波市亭下水库、晈口水库、横山水库和白溪水库。废水样品7个,采自浙江省杭州市萧山区某采矿场周围水域。
水样使用100 mL棕色玻璃瓶采集,于4℃冷藏运输、保存,6 h内检测。地表水样品使用一次性无菌注射器吸入约1.0 mL,经过0.2 μm有机相针式滤器过滤后注入液相色谱进样小瓶中待测。废水样品采用固相萃取净化,具体流程如下:用3 mL甲醇和3 mL水活化固相萃取小柱;将10 mL废水样品通过小柱,用5 mL 5%氨水淋洗小柱,弃去流出液;用10 mL含2%(v/v)甲酸的甲醇溶液将目标物从小柱中洗脱出来,收集洗脱液,取1 mL待测。
1.3 色谱条件
柱温40℃;流速0.25 mL/min;进样量5.0 μL;分离时间 3 min。流动相:乙腈-水(90∶10,v/v)。苦味酸、苦氨酸的保留时间分别为0.86和1.13 min。苦味酸和苦氨酸混合标准溶液的色谱图见图1。
1.4 质谱条件
三重四极杆串联质谱多反应监测(MRM);电喷雾电离(ESI)正离子电离模式;毛细管电压3.0 kV;离子源温度100℃;脱溶剂气温度200℃;脱溶剂气流量450 L/h;锥孔气流量50 L/h,各目标化合物的质谱分析参数见表1。
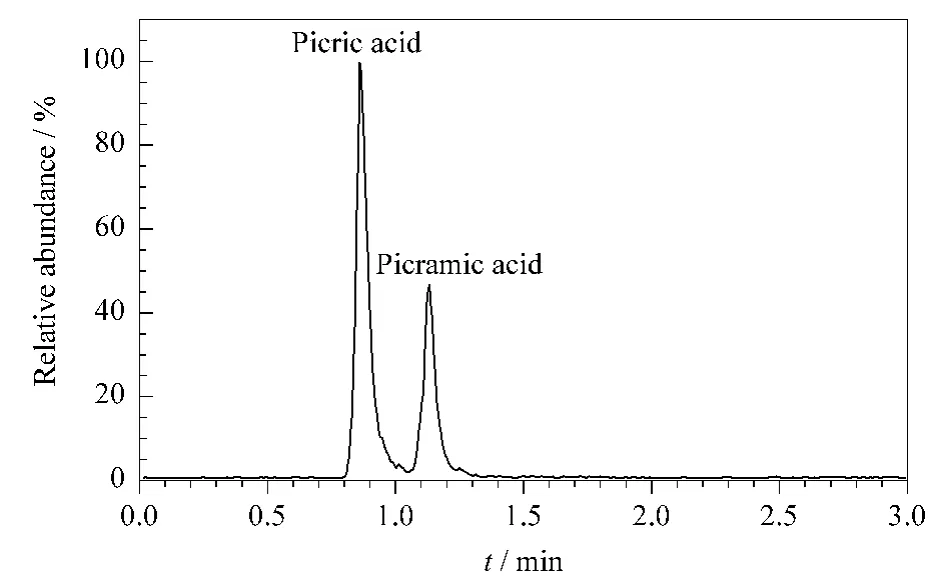
图1 苦味酸和苦氨酸混合标准溶液的HILIC色谱图Fig.1 HILIC chromatogram of picric acid and picramic acid
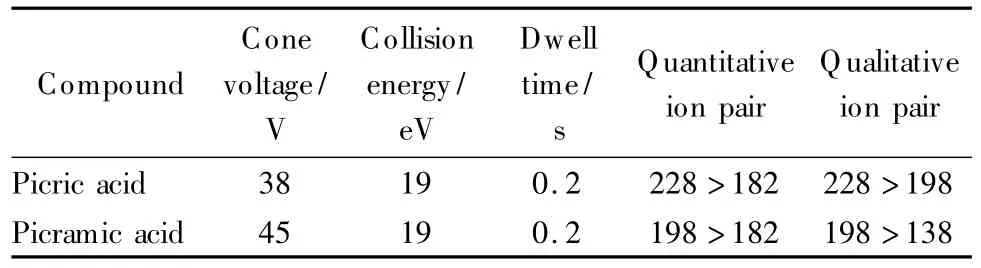
表1 苦味酸、苦氨酸的LC-MS参数Table 1 LC-MS parameters of picric acid and picramic acid
2 结果与讨论
2.1 色谱柱的选择
亲水色谱柱是近年来发展起来的针对强极性化合物的新型液相色谱柱。本文对比了反相C18色谱柱(Acquity UPLC BEH C18 column,100 mm ×2.1 mm,1.7 μm)和亲水作用色谱柱(Acquity UPLC BEH HILIC column,100 mm ×2.1 mm,1.7 μm)对苦味酸的保留,结果见图2。
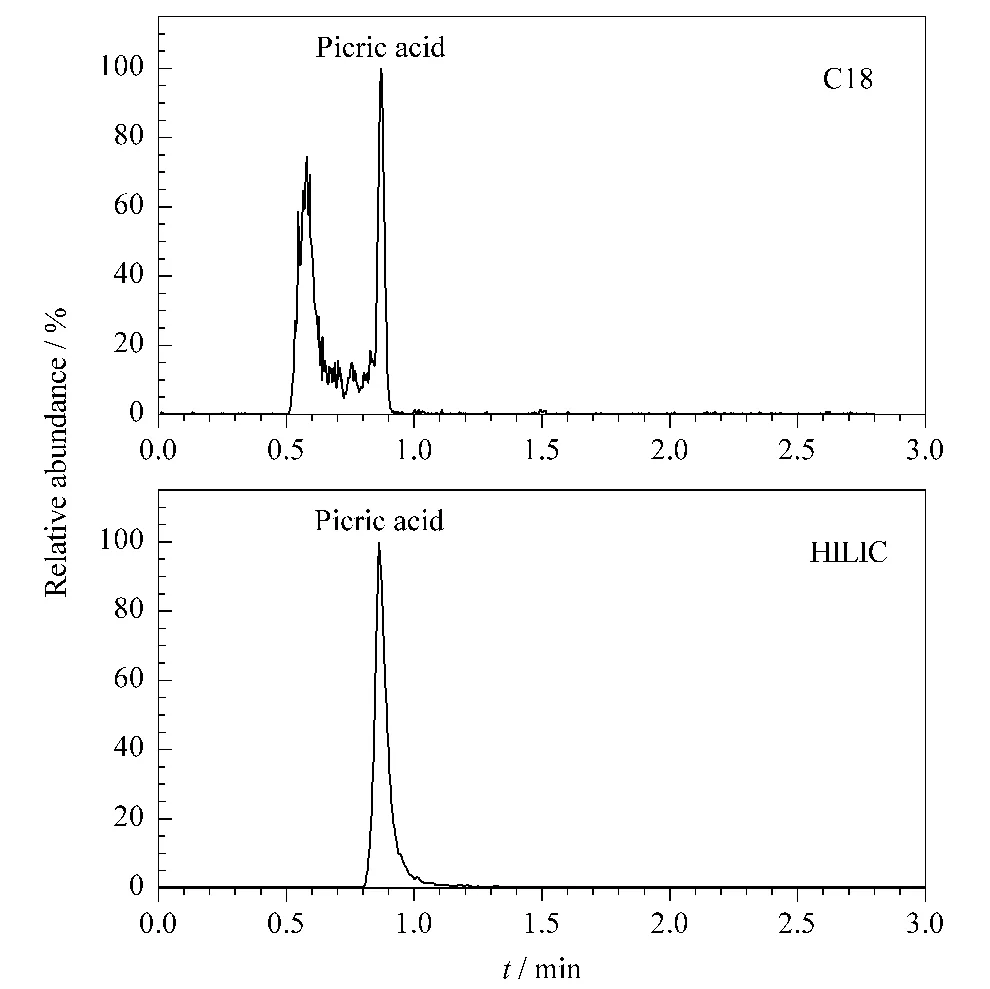
图2 不同色谱柱分析苦味酸的UPLC-MS/MS色谱图Fig.2 UPLC-MS/MS chromatograms of picric acid with different columns
从图2可以看出,在HILIC色谱柱中,采用乙腈/水体系分析苦味酸,不仅实现了强极性化合物的保留,而且灵敏度极高,峰形良好,无须使用缓冲盐;在同样的等度流动相情况下,苦味酸标准物质在C18色谱柱中出现一个很宽的延展峰,无法实现有效保留。
2.2 质谱条件的优化
质谱条件的优化是通过在流动注射模式下对1.0 mg/L的两种物质单独进样完成的。首先比较了ESI和大气压化学电离(APCI)两种电离源。在SIM模式下,ESI中两种化合物的灵敏度明显高于APCI,这与ESI更适用于极性物质的结论相符[11]。在MRM模式中,苦味酸和苦氨酸最主要的子离子都是m/z 181.7,对于苦味酸是[M -H -NO2]-,对于苦氨酸是[M -OH]-,二级质谱图见图3。
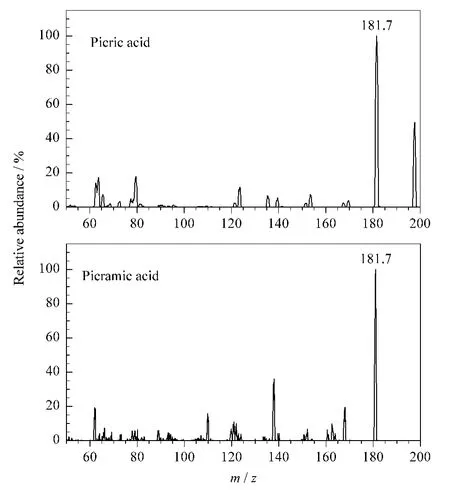
图3 苦味酸和苦氨酸的二级质谱图Fig.3 MS/MS spectra of picric acid and picramic acid
2.3 前处理条件的优化
在对废水样品进行处理时,选取了适合强酸性物质的Oasis MAX固相萃取小柱进行萃取。由于本方法的检测灵敏度足够高,本研究未进行复杂的样品浓缩富集,简化了样品前处理流程,既缩短了检测时间,又提高了检测准确度。在低、中、高3种浓度水平下同时添加两种化合物标准品(加标质量浓度为 1.0、10.0、100.0 μg/L),在地表水样品中的回收率为89%~107%,在废水样品中的回收率为72%~101%,相对标准偏差不超过20%。
2.4 方法验证
实验室空白和方法空白均未检出目标化合物。以2.0、20.0、100.0 μg/L空白加标溶液做6次平行试验,相对标准偏差为4.9%~14.7%。以空白实际水样为基质,添加标准溶液,通过低浓度曲线外推法确定检出限(三倍信噪比),苦味酸和苦氨酸的检出限分别为0.1和0.3 μg/L,满足国家地表水标准限值要求[12]。
2.5 实际样品检测
在15个地表水样品中均未检出苦味酸和苦氨酸,在6个采矿场周围水域样品中苦味酸检测结果为未检出~59.5 μg/L;苦氨酸检测结果为未检出~12.3 μg/L。
3 结论
本研究采用HILIC色谱柱实现了强极性物质苦味酸在液相色谱中的有效保留。地表水样品直接进样即可检测,废水样品也只需通过SPE净化而无需浓缩。串联质谱法对苦味酸和苦氨酸定性准确。本方法操作简便,选择性好,灵敏度高,分析速度快,适用于地表水、生活污水、工业废水等多种实际水样的检测。
[1] Astratov M,Preiβ A,Levsen K,et al.Int J Mass Spectrom Ion Proc,1997,167/168:481
[2] Rajan J,Valli K,Perkins R E,et al.J Ind Microbiol,1996,16(5):319
[3] Kumar M,Reja S I,Bhalla V.Org Lett,2012,14(23):6084
[4] GB/T 5750.8-2006
[5] Fallas M M,Tanaka N,Buckenmaier S M C,et al.J Chromatogr A,2013,1297:37
[6] Van Nuijs A L N,Tarcomnicu I,Covaci A.J Chromatogr A,2011,1218(35):5964
[7] Pamme N,Steinbach K,Ensinger W J,et al.J Chromatogr A,2001,943(1):47
[8] Nipper M,Qian Y,Carr R S,et al.Chemosphere,2004,56(6):519
[9] Yost S L,Pennington J C,Brannon J M,et al.Mar Pollut Bull,2007,54(8):1262
[10] Guo Y L,Yuan Q,Li R P,et al.Chinese Journal of Chromatography(郭亚丽,袁琴,李瑞萍,等.色谱),2012,30(3):232
[11] Reemtsma T.J Chromatogr A,2003,1000(1/2):477
[12] GB 3838-2002
猜你喜欢
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:56:34
现代仪器与医疗(2021年1期)2021-06-09 05:53:38
煤矿爆破(2021年1期)2021-06-03 07:43:22
火炸药学报(2019年2期)2019-05-05 08:33:54
净水技术(2018年3期)2018-04-02 11:44:28
中国资源综合利用(2017年4期)2018-01-22 02:46:54
广东饲料(2016年7期)2016-12-01 03:43:36
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
江苏调味副食品(2015年1期)2015-02-28 01:56:34
食品工业科技(2014年23期)2014-03-11 18:18:54
