
超高效液相色谱-串联质谱法测定动物源性食品中的磺胺增效剂
2014-05-08 11:14高洋洋张朝晖卢晓宇杨大进
色谱 2014年5期
高洋洋, 张朝晖, 刘 鑫, 卢晓宇, 严 华,何 悦, 杨大进, 云 环*
(1.北京出入境检验检疫局技术中心,北京100026;2.国家食品安全风险评估中心,北京100021)
磺胺增效剂是指含有5-取代苄基-2,4-二氨基嘧啶的一类化合物,包括三甲氧苄氨嘧啶(trimethoprim,TMP)、二甲氧苄胺嘧啶(diaveridine,DVD)和二甲氧甲基苄胺嘧啶(ormetoprin,OMP)等(结构见图1),为广谱抑制剂,多与磺胺合用使细菌的叶酸代谢受到双重阻断,抗菌作用可增效数十倍。磺胺增效剂常用于畜禽养殖业中治疗大肠杆菌引起的败血症、鸡白痢及球虫病等[1]。由于其在动物体内代谢缓慢,残留在肌肉组织及器官中的时间较长,并可通过食物链传递,人食用后会产生毒副作用及抗药性。各国对磺胺增效剂都有严格的控制法规:日本肯定列表对鱼类中磺胺增效剂限量要求为50 ~100 μg/kg[2];欧盟 1181/2002/EC 中也明确规定其不可用于产蛋类禽养殖业[3];我国农业部对磺胺增效剂的使用也明确规定三甲氧苄氨嘧啶限量不得超过 50 μg/kg[4]。

图1 三甲氧苄氨嘧啶(TMP)、二甲氧苄氨嘧啶(DVD)、二甲氧甲基苄氨嘧啶(OMP)的结构式Fig.1 Chemical structures of TMP,DVD and OMP TMP:trimethoprim;DVD:diaveridine;OMP:ormetoprin.
测定磺胺增效剂的方法主要有荧光分光光度法[5]、示差分光光度法[6]、胶体金免疫层析法[7]、液相色谱法[8-11]和液相色谱-串联质谱法[12-14]等。荧光分光光度法的选择性较差,示差分光光度法易受干扰物的影响,胶体金免疫层析法的重复性较差,液相色谱法的灵敏度较低且无法提供结构信息。液相色谱-串联质谱法可提供待测物的结构信息,具有较高的灵敏度和选择性,是同时测定上述3种组分的理想方法。动物源性食品种类多、范围广,但有关磺胺增效剂检测的报道所涉及的基质种类较少,多集中在肉类产品[15],乳制品仅有少量文献[16-18]报道,对复杂基质中磺胺增效剂的测定需要单独建立前处理方法。本实验不仅针对肉类和乳制品,还针对含有大量蛋白质、脂肪及核黄素的鸡肝和含有特殊磷脂质的鸡蛋提供了一种统一的、有效的前处理方法,和现有文献方法相比,适用性更为广泛且简便快速,极大地节省了检测时间和检测成本。
本文针对鸡肉、鱼肉、鸡肝、鸡蛋、牛奶等多种基质采用甲酸-乙腈作提取溶液,正己烷一步除脂,BEH C18柱色谱分离后经超高效液相色谱-串联质谱(UPLC-MS/MS)测定,建立了磺胺增效剂的准确定性、定量检测方法,具有高灵敏度和可靠性,结果令人满意。
1 实验部分
1.1 仪器与试剂
UPLC-Xevo TQ MS型超高效液相色谱-串联质谱仪(美国Waters公司);Milli-Q超纯水系统(美国Millipore公司);微孔滤膜(0.2 μm,美国Pall公司);超声波清洗器(江苏昆山超声仪器有限公司);小型高速离心机(德国Sigma公司);Aspecxl多通道固相萃取仪(法国Gilson公司);PCX固相萃取小柱(3 mL,美国菲罗门公司)。
三甲氧苄氨嘧啶(纯度98.7%)、二甲氧苄氨嘧啶(纯度99%)、二甲氧甲基苄氨嘧啶(纯度99%)标准品(德国Dr.Ehrenstorfer公司)。三氯乙酸、氨水(色谱纯,美国TEDIA公司);乙腈、甲醇、甲酸、正己烷、醋酸铵(色谱纯,美国Fisher公司);二氯甲烷(分析级,国药集团化学试剂有限公司);实验用水均由Milli-Q超纯水系统制备。
1.2 标准溶液的配制
标准储备液:准确称取三甲氧苄氨嘧啶、二甲氧苄氨嘧啶和二甲氧甲基苄氨嘧啶标准品用甲醇分别配制质量浓度为100 mg/L的标准储备液,于4℃下避光保存。
混合标准中间液:取上述标准储备液用甲醇配制成质量浓度为1 mg/L的混合标准中间溶液,于4℃下避光保存。
1.3 UPLC-MS/MS 条件
1.3.1 UPLC 条件
色谱柱:Acquity UPLC BEH C18色谱柱(50 mm×2.1 mm,1.7 μm;美国 Waters公司)。流动相 A为甲醇,流动相B为5 mmol/L醋酸铵(含0.1%(v/v)甲酸)。梯度洗脱程序:0~1.0 min,10%A~20%A;1.0~2.0 min,20%A ~70%A;2.0~3.5 min,70%A~80%A;3.5~4.2 min,80%A ~10%A。流速:0.25 mL/min;柱温:35 ℃;进样体积:10.0 μL。
1.3.2 MS/MS 条件
电喷雾离子源,正离子(ESI+)模式;检测方式:多反应监测(MRM);电喷雾电压:3.6 kV;雾化气温度:400℃;监测离子对、锥孔电压和碰撞能量等参数见表1。

表1 3种磺胺增效剂的多反应监测模式的串联质谱参数Table 1 MS/MS parameters in multiple reaction monitoring(MR M)mode for the three sulfonamide potentiators
1.4 样品提取和净化
1.4.1 基质分散法(适用于鸡肉、鱼肉、蛋、鸡肝、脱脂牛奶,稀释倍数为2.5)
准确称取2 g(精确到0.01 g)均匀样品于干净的50 mL塑料离心管中,加入5 g无水硫酸钠,混匀后加入10 mL甲酸-乙腈(1∶9,v/v)提取液,涡漩混匀,超声提取20 min,离心10 min(8 000 r/min),取上清液2 mL置于10 mL离心管中待净化。
取待净化溶液,于35℃下浓缩至干。加入1.00 mL水溶解残渣,加入3 mL正己烷涡漩混匀2 min,于5 000 r/min下离心3 min,将下层液过0.2 μm滤膜,滤液供UPLC-MS/MS分析。
1.4.2 固相萃取法(适用于全脂牛奶等含乳量高的样品,稀释倍数为1)
准确称取2 g(精确至0.01 g)均匀样品于干净的50 mL塑料离心管中,加入10 mL乙腈,涡漩混匀,超声提取20 min,离心10 min(8 000 r/min),取上清液5 mL到10 mL离心管中待净化。
将待净化溶液移入PCX固相萃取柱(使用前用3 mL水和3 mL甲醇活化),用3 mL水和3 mL甲醇淋洗,用3 mL氨水-甲醇(1∶19,v/v)洗脱,于35℃下浓缩至干。加入1.00 mL水溶解残渣,过0.2 μm滤膜,滤液供UPLC-MS/MS分析。
2 结果与讨论
2.1 前处理条件的优化
2.1.1 提取方法的优化
本实验以鸡肉、鱼肉、鸡肝、鸡蛋、牛奶样品的加标回收试验(加标水平为10 μg/kg,平行测定3份)考察了乙腈、甲酸-乙腈(1∶9,v/v)、三氯乙酸-乙腈(7∶3,v/v)3种提取溶液对三甲氧苄胺嘧啶的提取效率,发现甲酸-乙腈(1∶9,v/v)可获得较高的提取效率(见表2)。这是由于磺胺增效剂结构中含有多个氨基,呈强极性,难溶于非极性溶剂,易溶于极性有机溶剂,同时充分利用了乙腈能使蛋白质变性沉淀的作用,简化了实验过程,而甲酸可使磺胺增效剂在酸性条件下更易形成正离子。
另外,采用基质分散法提取样品时加入适量的无水硫酸钠不仅可以防止样品结块,同时还可以吸收样品中的水分及干扰成分以免影响提取效率。
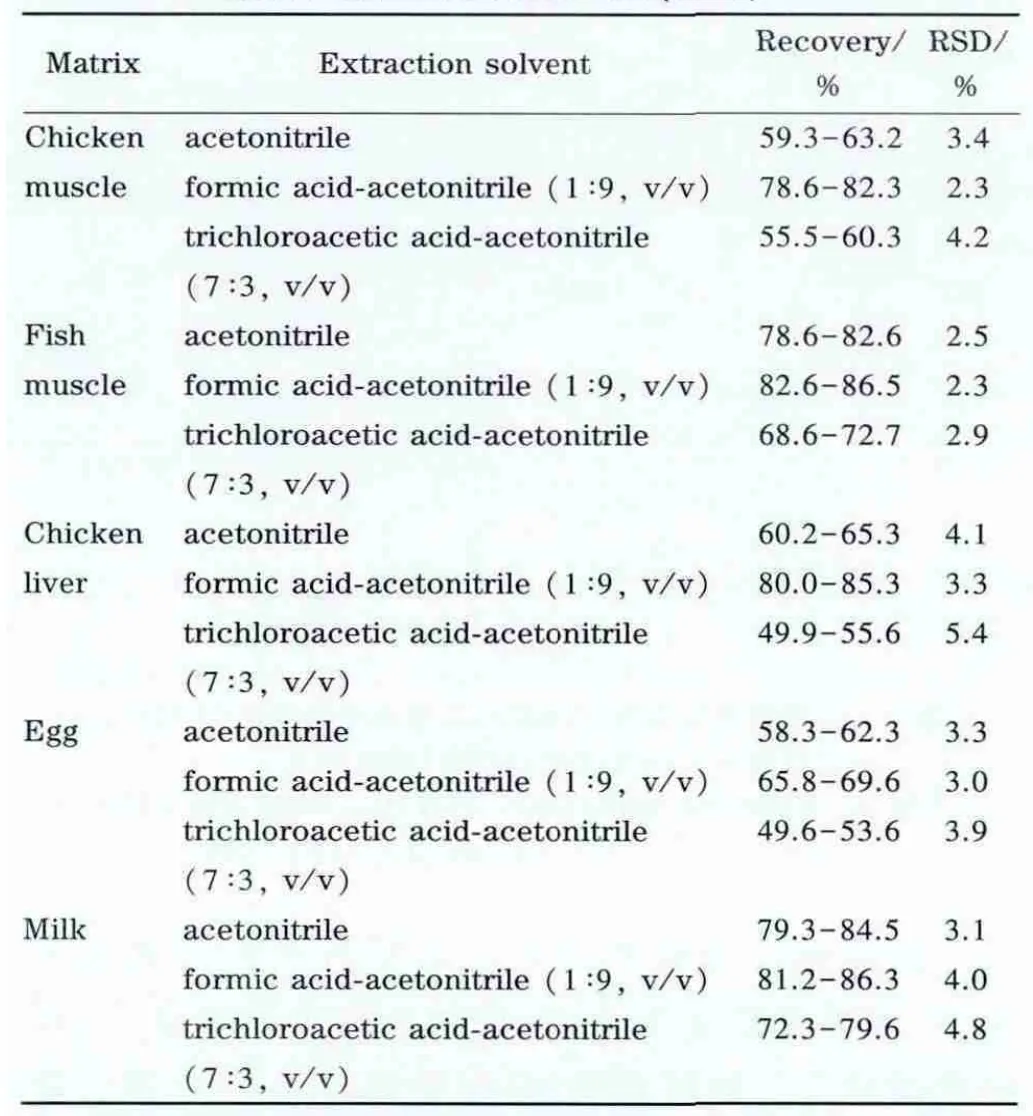
表2 不同提取液对鸡肉、鱼肉、鸡肝、鸡蛋和牛奶样品中三甲氧苄胺嘧啶的提取回收率和相对标准偏差(n=3)Table 2 R ecoveries and relative standard deviations of trimethoprim in chicken muscle,fish muscle,chicken liver,egg and milk matrices with different extraction solvents(n=3)
2.1.2 净化条件的优化
实验初期尝试以二氯甲烷为除脂溶剂,发现二氯甲烷除脂后的回收率仅为19.68%~32.00%,这是由于二氯甲烷在除脂的同时也溶解了一部分目标分析物;改用正己烷除脂,回收率可达到90.65%~105.05%。
由于鸡肉、鱼肉、鸡肝和鸡蛋基质内含有的脂肪较高,本文分别尝试了用正己烷2 mL一次除脂、2 mL二次重复除脂和3 mL一次除脂。实验结果显示:用3 mL正己烷一次除脂最为理想;用2 mL正己烷一次除脂后样品液中脂肪层过厚、净化液剩余量过少;用2 mL正己烷二次除脂和3 mL正己烷一次除脂的效果相当,但二次除脂操作复杂耗时长。将该方法应用于脱脂牛奶基质的前处理,其净化效果和回收率也可达到实验要求。
2.1.3 浓缩条件的优化
磺胺增效剂具有热不稳定性,浓缩时如果高温加热,虽能节省时间,但化合物被分解,回收率降低,得不偿失,故加热温度不能设定太高。乙腈由于沸点较高,蒸发耗时较长,在温度较低的情况下耗时更长。通过试验比较,本实验选用35℃为浓缩最佳温度,既满足回收率要求又最大限度地节省了时间。浓缩时不能过分蒸干,要及时观察蒸发状态,蒸发完毕后马上定容,以免提取液浓缩后黏附于试管壁上难以溶解,导致回收率下降。
2.1.4 固相萃取净化柱的选择
本实验以全脂牛奶等含乳量高的样品的加标回收试验(加标量为10 ng/mL)考察了PCX和MCX(美国Waters公司)、SCX(美国菲罗门公司)、HLB(美国Waters公司)4种固相萃取柱的净化效果和回收效率。结果(见表3)显示:PCX柱的保留较好,回收率良好。这是由于PCX柱是以聚苯乙烯-二乙烯苯为填料的萃取柱,该填料在酸性、中性、碱性中都很稳定,属于水可浸润型聚合物,该萃取柱采用离子交换与反相保留双重保留模式实现了良好的净化效果和回收效率。而待测组分在HLB柱中基本不保留;在MCX、SCX柱中部分保留,回收率较低。故最终选用PCX作为固相萃取净化柱。

表3 不同固相萃取柱对全脂牛奶中磺胺增效剂的回收率和相对标准偏差(n=3)Table 3 R ecoveries and relative standard deviations of the sulfonamide potentiators in milk on different solid phase extraction columns(n=3)
2.2 UPLC条件的优化
2.2.1 流动相的选择
选择甲醇为流动相的有机相是因为甲醇与乙腈相比极性较弱,当流动相梯度条件相同时,甲醇可使3种磺胺增效剂的保留时间相对推后,同时甲醇会提高离子化程度,峰面积比以乙腈为流动相时大。选择醋酸铵为流动相的水相可增加磺胺增效剂的保留性;加入少量甲酸可促进电离,提高灵敏度。通过试验比较,本实验选择流动相A为甲醇,B为5 mmol/L醋酸铵(含0.1%(v/v)甲酸)。
2.2.2 柱温的选择
考察了柱温分别为35、40和45℃时3种磺胺增效剂灵敏度的差别,结果显示随着柱温的升高,检测灵敏度无明显变化。考虑到色谱柱在相对较低温度下使用寿命较长,本实验设定柱温为35℃。
2.3 UPLC-MS/MS 分析
在优化的UPLC-MS/MS条件下三甲氧苄氨嘧啶、二甲氧苄氨嘧啶和二甲氧甲基苄胺嘧啶的保留时间为2.23、2.61和2.88 min。鸡肉样品中添加10.0 μg/kg 3种磺胺增效剂的MRM 谱图见图2。
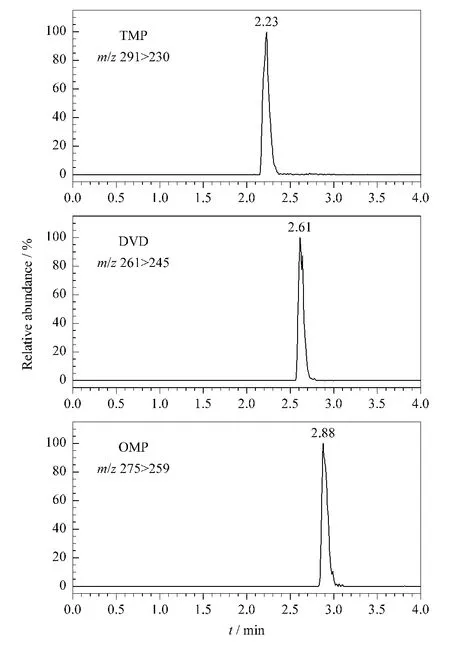
图2 加标(10.0 μg/kg)鸡肉样品的UPLC-MS/MS MR M谱图Fig.2 Chromatograms of a chicken sample spiked with the sulfonamide potentiator standards at 10.0 μg/kg by UPLC-MS/MS in MR M mode
2.4 基质效应、标准曲线和定量限(LOQ)
实验发现待测组分在基质中受到的基质干扰非常明显。取空白样品按1.4节进行前处理,以此基质提取液作稀释液配制混合标准溶液,与直接用定容液配制的标准溶液对比,基质增强效率约为6倍。为扣除此基质效应,本实验采用空白样品配制基质标准曲线。分别取不同基质的空白样品进行前处理,以基质提取液作稀释液配制混合标准曲线,以定量离子的峰面积对质量浓度进行线性回归,在1.25~30.0 μg/L范围内线性关系良好(r≥0.99)。在空白样品中添加不同浓度的待测组分,前处理后按浓度由高至低检测,直到获得信噪比等于10(S/N=10)的浓度,确定定量限(LOQ)为 5.0 μg/kg。
2.5 方法的回收率和精密度
表4为在空白试样中进行的添加回收率和精密度测定结果。在鸡肉、鱼肉、鸡肝、鸡蛋及牛奶样品中分别添加 5.0、10.0、20.0 μg/kg的混合标准溶液,混合后静置,使标准溶液被样品充分吸收,然后按照本方法测定,每个添加水平测定6次,计算加标回收率和相对标准偏差(RSD)。
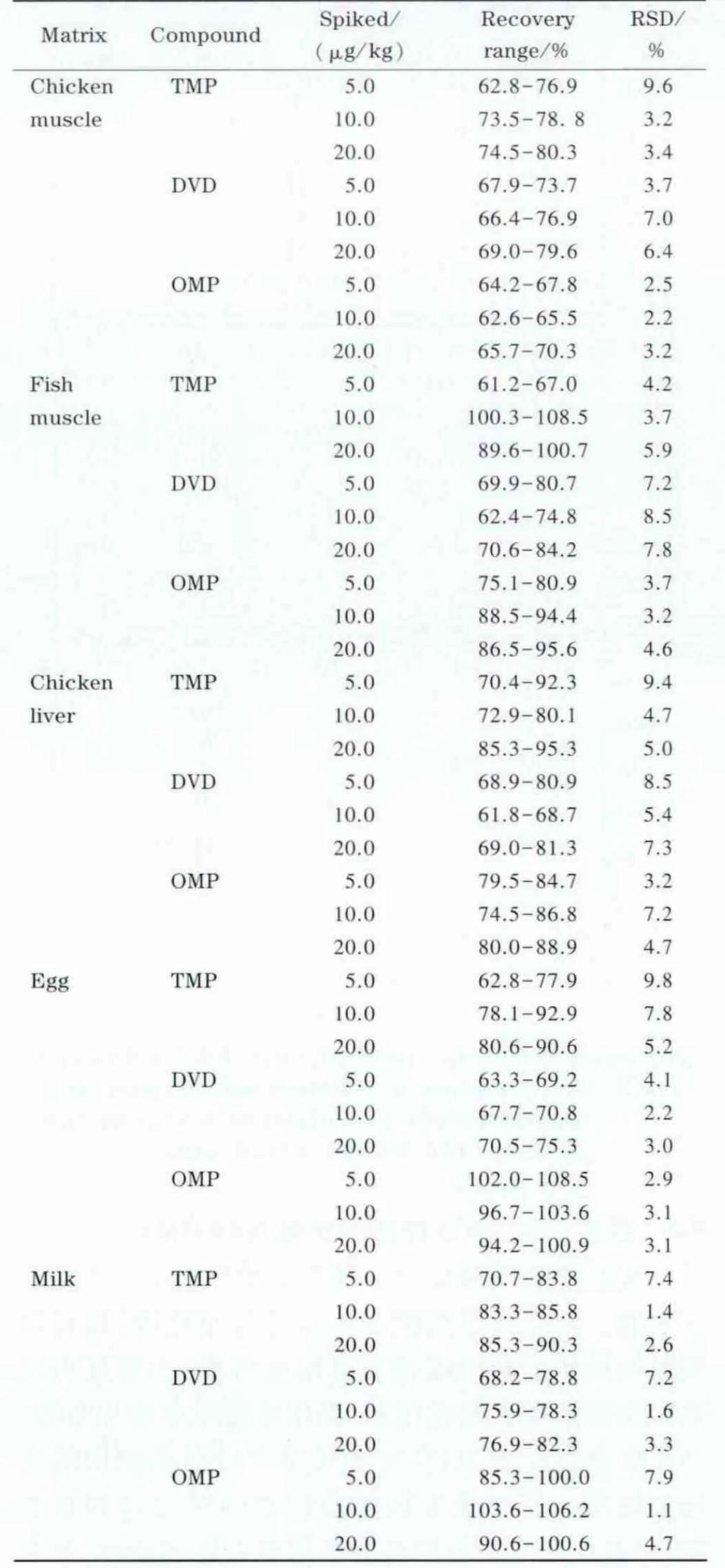
表4 各种基质中3种磺胺增效剂的加标回收率范围和R SD(n=6)Table 4 R ecovery ranges and R SDs of the three sulfonamide potentiators spiked in different matrices(n=6)
3 结论
本文建立了一种快速有效的多种动物源性食品中3种磺胺增效剂的超高效液相色谱-串联质谱检测方法,获得了满意的分离效果和检测灵敏度。该方法针对鸡肉、鱼肉、鸡肝、鸡蛋和牛奶等基质尝试了统一、简单的前处理方法,对比固相萃取的净化方式,在保证净化效果的同时有效地提高了回收率。该方法的回收率、精密度和定量限均满足残留分析的要求。
[1] Song W,Hu Y Y,Han F,et al.Chinese Journal of Chromatography(宋伟,胡艳云,韩芳,等.色谱),2013,31(12):1161
[2] Li B,Cheng H,Zhan C R.Physical Testing and Chemical Analysis Part B:Chemical Analysis(李兵,程慧,占春瑞.理化检验:化学分册),2010,46(11):1348
[3] Commission Regulation(EC)No.1181/2002
[4] NY 5070-2002
[5] Gao J H,Lin P,Chen B.Analytical Laboratory(高建华,林鹏,陈彬.分析试验室),1999,18(6):37
[6] Li H Y,Fu G H.Shandong Pharmaceutical Industry(李华玉,伏光华.山东医药工业),1996,15(3):21
[7] Wan Y P,Feng C W,Zhao Z M,et al.China Dairy Industry(万宇平,冯才伟,赵正苗,等.中国乳品工业),2013,41(2):24
[8] Liu Z W,Jiang Z X,Cao X,et al.China Animal Quarantine(刘振伟,姜兆兴,曹旭,等.中国动物检疫),2010,27(5):36
[9] Cheng L L,Zhang S X,Shen J Z,et al.Journal of Instrumental Analysis(程林丽,张素霞,沈建忠,等.分析测试学报),2008,27(1):88
[10] GB/T 21037-2007
[11] Sayar E,Sahin S,Cevheroglu S,et al.Eur J Drug Metab Ph,2011,35(12):41
[12] Du Y,Yang H Y,Xu W D.Journal of Pharmaceutical Analysis(杜玥,杨慧元,徐伟东.药物分析杂志),2010,30(3):471
[13] Guo W,Liu Y,Liu N,et al.Chinese Journal of Analytical Chemistry(郭伟,刘永,刘宁,等.分析化学),2009,37(11):1638
[14] Jiang Y,Shen C Y,Yao Y G,et al.Journal of Instrumental A-nalysis(蒋原,沈崇钰,姚义刚,等.分析测试学报),2009,28(7):834
[15] Li H,Sun H W,Zhang J X,et al.J Food Control,2013,31:359
[16] GB/T 22966-2008
[17] SN/T 2312-2009
[18] Wu Y L,Zhao L,Liu Y J,et al.Chinese Journal of Chromatography(吴银良,赵莉,刘勇军,等.色谱),2007,25(5):728
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
今日农业(2020年16期)2020-09-25
天津农林科技(2020年3期)2020-08-13
中国化肥信息(2020年7期)2020-03-19
武警医学(2018年10期)2018-11-06
当代化工研究(2016年6期)2016-03-20
湖南农业(2016年12期)2016-03-10
无机化学学报(2014年3期)2014-02-28
中国兽药杂志(2012年4期)2012-11-06
