
液相色谱-串联质谱法测定面粉及其制品中的联二脲
2014-05-08 11:14:54王俊苏郗存显陈冬东王国民母昭德
色谱 2014年5期
王 雅, 王俊苏, 向 露, 郗存显, 陈冬东,彭 涛, 王国民, 母昭德*
(1.重庆医科大学药学院,重庆400016;2.重庆出入境检验检疫局,重庆市进出口食品安全工程技术研究中心,重庆400020;3.中国检验检疫科学研究院,北京100123)
2014年2月,有外媒报道称赛百味、麦当劳、星巴克、汉堡王等使用添加了偶氮甲酰胺(azodicarbonamide,ADA)的面粉来制作面包,该成分通常作为橡胶鞋底和瑜伽垫的原料,这一消息引发了消费者对面粉类食品安全的担心。ADA作为食品添加剂在面粉中广泛使用,具有氧化与漂白双重作用,其主要目的是用来增加面筋,改善面团流变学特性和机械加工性能[1]。由于ADA的降解产物氨基脲(semicarbazide,SEM)具有较强的致突变和致癌作用[2],我国 GB2760-2011《食品添加剂使用卫生标准》规定小麦粉中ADA的最大使用量为45 mg/kg[3]。近年来,有大量研究表明ADA在面制品的加工过程中会全部降解,生成联二脲(biurea,HDC)与SEM 残留于面制品中[4-7]。由于在面制品中检测不到ADA的存在,因此有必要建立面粉及制品中ADA的特征降解产物HDC的检测方法,从而有效地监控面粉中ADA的使用。
目前,国内外关于HDC的分析方法主要有红外光谱法[8]、核磁共振法[8]、高效液相色谱法(HPLC)[9]和 液 相 色 谱-串 联 质 谱 法 (LC-MS/MS)[10,11]。红外光谱和核磁共振分析只适合于对工业上生产的HDC进行结构表征。与LC-MS/MS比较,HPLC的定性分析能力较差,无法对HDC进行准确的定性分析,容易出现假阳性,而且灵敏度较低。Mulder等[10]利用LC-MS/MS建立了面粉处理的肉制品中HDC的测定方法,袁丽红等[11]建立了面粉中HDC的LC-MS/MS测定方法,而对于面粉制品中HDC的研究报告却很少。由于HDC在串联质谱中响应较低,直接测定HDC有难度,因此本研究建立了HDC的衍生化法,对面粉及多种制品中的HDC进行了测定及确证:采用高锰酸钾将HDC氧化生成ADA,利用对甲苯亚磺酸钠对ADA进行衍生化,生成对甲苯磺酰氨基脲,最后采用LCMS/MS进行测定和确证。衍生化原理如图1所示[12,13]。该方法新颖、灵敏度高、定性定量准确且测定范围宽,适用于面粉及多种面粉制品,可以为面粉及其制品中HDC的分析提供技术支持。

图1 HDC的衍生化反应原理Fig.1 Principle of HDC derivatization
1 实验部分
1.1 仪器、试剂与材料
API4000高效液相色谱-串联质谱仪(美国AB-sciex公司);SR-2DW 型强力振荡器(北京安捷来科学仪器设备有限公司);3-30K型离心机(德国Sigma公司);XH-B型旋涡混合器(江苏康健医疗用品有限公司);Sartorius BS224S型天平(赛多利斯科学仪器有限公司);Milli-Q超纯水系统(美国Millipore公司);AS 10200B型超声水浴仪(天津奥特赛恩斯仪器有限公司)。
联二脲标准品(纯度≥99.5%,美国Chem Service);13C2,15N2-联二脲(加拿大 Toronto Research Chemicals);邻硝基苯甲醛(纯度为98%,美国Sigma公司);水为超纯水;亚铁氰化钾(成都市科龙化工试剂厂);乙酸锌(上海强顺化工试剂有限公司);高锰酸钾(重庆川东化工有限公司);乙腈和二甲基亚砜(HPLC级,美国Tedia公司);冰醋酸(HPLC级,美国Fluka公司);甲酸(纯度>99%,美国ROE公司);对甲苯亚磺酸钠(纯度>95%,美国Sigma公司),乙酸铵(纯度>99%,美国Sigma公司)。面粉、馒头、面包、方便面、油条均为市售。
1.2 实验条件
1.2.1 溶液的配制
标准储备液:分别精密称取适量HDC标准品(精确至0.01 mg),用水配制成质量浓度为 20 mg/L的标准储备液;置于室温保存备用,有效期30 d;根据需要,用水稀释成相应质量浓度的标准工作溶液。内标工作液:分别精密称取适量HDC内标物质(精确至0.01 mg),用水配制成质量浓度为10 mg/L的标准溶液;置于室温保存备用,有效期30 d。0.25 mol/L亚铁氰化钾溶液:称取106.0 g亚铁氰化钾,用水溶解并稀释至1 L;室温保存,有效期90 d。1.00 mol/L乙酸锌溶液:称取220.0 g乙酸锌,加30 mL冰醋酸溶解,用水稀释至1 L;室温保存,有效期90 d。0.06 mol/L高锰酸钾溶液:称取高锰酸钾1 g,用水溶解并稀释至100 mL;室温保存,有效期90 d。2.5 mmol/L对甲苯亚磺酸钠溶液:称取0.043 5 g对甲苯亚磺酸钠,加入50%乙腈水溶液溶解并稀释至100 mL;室温保存,有效期30 d。2.0 mmol/L乙酸铵水溶液(含0.2%甲酸):称取0.154 2 g乙酸铵溶解于900 mL的水中,加入2 mL甲酸,用水定容至1 L,超声脱气。
1.2.2 色谱条件
色谱柱:Shimpak XR-ODSⅡ色谱柱(150 mm×2.0 mm,2.2 μm);柱温:40 ℃;进样体积:10 μL。流动相:A为乙腈,B为2 mmol/L乙酸铵溶液(含0.2%(v/v)甲酸),流速0.3 mL/min。梯度洗脱程序:0~2 min,5%A;2~4 min,5%A~90%A;4~7 min,90%A;7~7.5 min,90%A ~5%A;7.5~10 min,5%A。
1.2.3 质谱条件
离子源:电喷雾离子源(ESI);离子化模式:正离子模式;扫描方式:多反应监测(MRM)模式;电喷雾电压(IS):5 500 V;雾化气压力(GS1):551.6 kPa;气帘气压力(CUR):241.3 kPa;辅助气压力(GS2):344.7 kPa;离子源温度(TEM):650℃;碰撞室入口电压(EP):10 V;碰撞室出口电压(CXP):12 V;监测离子对、驻留时间、去簇电压、碰撞电压等参数见表1。

表1 待测物的离子对、驻留时间、去簇电压和碰撞电压Table 1 Ion pairs,dwell times,declustering potentials and collision voltages of the analytes
1.2.4 样品预处理
准确称取2.0 g试样于50 mL离心管中,分别加入0.2 mL HDC内标溶液和10 mL水,旋涡振荡1 min后,置于振荡器中振荡15 min,于8 000 r/min的转速离心5 min,移取上清液于25 mL比色管中;残留物再用10 mL水提取1次;合并提取液,分别加入2 mL乙酸锌溶液与亚铁氰化钾溶液,加水定容至刻度,振荡混匀;溶液转移至50 mL离心管,再加入5 mL正己烷脱脂,于8 000 r/min的转速离心3 min;准确移取5 mL下层清液于另一离心管中,加入0.06 mol/L高锰酸钾溶液200 μL,振荡反应30 min后,加入50 μL二甲基亚砜,振荡3 min后,加入2.5 mmol/L对甲苯亚磺酸钠溶液5 mL,振荡衍生10 min,离心,过0.22 μm 有机滤膜后供LC-MS/MS测定。
2 结果与讨论
2.1 LC-MS/MS测定条件的优化
将待测化合物(对甲苯磺酰氨基脲)溶液以流动注射的方式注入电喷雾离子源,分别以正离子和负离子模式进行扫描,发现在正离子模式下分子离子[M+H]+m/z 230.0响应值较高,因此采用正离子扫描方式。在ESI+模式下对分子离子的子离子进行扫描,选取丰度较强、干扰较小的两个子离子m/z 157.0和m/z 97.3为监测离子,其中丰度最高的子离子m/z 157.0作为定量离子。再对碰撞能量、雾化气压力、电喷雾电压等参数进行优化,从而确定各子离子的最佳检测条件。对甲苯磺酰氨基脲及内标物母离子的二级质谱图见图2,定量离子提取离子色谱图见图3。
液相色谱分离时在流动相中加入适量甲酸和挥发性电解质乙酸铵可使离子预先形成,提高待测物在ESI+模式下的电离效率,从而提高检测的灵敏度。同时结合缓冲盐可以调节流动相pH值、保证良好的峰形和保持离子强度的特点,本实验考察了乙腈-0.2%甲酸溶液(分别含1.0、2.0、5.0和8.0 mmol/L乙酸铵)对目标物响应强度的影响。结果表明,采用乙腈-2 mmol/L乙酸铵溶液(含0.2%(v/v)甲酸)为流动相时,在1.2.2节的梯度洗脱条件下,目标物的峰形好且响应强度较大,分离度和灵敏度较高,因此选择乙腈-2 mmol/L乙酸铵溶液(含0.2%甲酸)作为流动相。

图2 (a)对甲苯磺酰氨基脲与(b)13C,15N-对甲苯磺酰氨基脲的二级质谱图Fig.2 MS/MS spectra of(a)p-toluenesulfonyl semicarbazide and(b)13C,15N-p-toluenesulfonyl semicarbazide
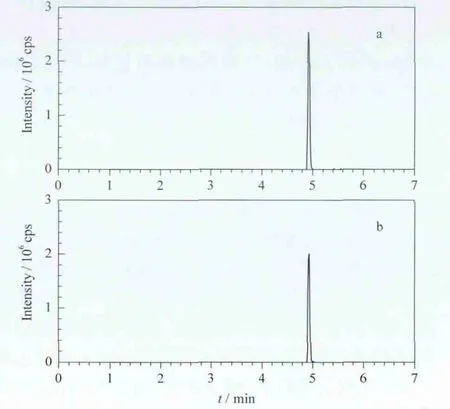
图3 (a)对甲苯磺酰氨基脲及(b)13C,15N-对甲苯磺酰氨基脲的定量离子流色谱图Fig.3 Quantitative ion current chromatograms of(a)ptoluenesulfonyl semicarbazide and(b)13C,15N-ptoluenesulfonyl semicarbazide
2.2 前处理条件的优化
2.2.1 提取溶剂的选择
HDC不溶于大部分有机溶剂,微溶于水、丙酮和 N,N-二 甲 基 甲 酰 胺 (dimethyl formamide,DMF)。Mulder等[10]采用 DMF-水(9∶1,v/v)溶液提取禽肉和海产品中的HDC,袁丽红等[11]则采用纯水提取面粉中的HDC。分别称取不含HDC的空白基质(面粉、馒头、面包、油条、方便面)2.0 g,分别添加10 mg/kg的HDC标准品,考察水、丙酮、DMF、丙酮-水(3∶7,v/v)、丙酮-水(1∶1,v/v)、丙酮-水(8∶2,v/v)、DMF-水(7∶3,v/v)、DMF-水(1∶1,v/v)、DMF-水(3∶7,v/v)、DMF-水(5∶95,v/v)、DMF-丙酮-水(1∶1∶2,v/v/v)的提取能力。结果(见表2)表明,采用纯水提取时,测得的HDC的含量较高。因此,本研究采用纯水作为提取溶剂。
2.2.2 提取方式与时间的优化
本研究分别考察了振荡提取、超声提取与涡旋振荡3种提取方式对试样的提取效果,发现振荡提取法的提取效果较好。另外比较了10 mL水提取多次的提取效果。结果表明,10 mL水提取2次后,增加提取次数,回收率增加较小,因此将提取次数定为2次。
2.2.3 衍生化条件的选择和优化
采用LC-MS/MS测定时,HDC在串联质谱中的响应较低。为了提高其检测灵敏度,需要对其进行衍生化。HDC与SEM的结构相似,而SEM的测定选用邻硝基苯甲醛作为衍生化试剂[14],本研究考察了邻硝基苯甲醛对HDC的衍生化效果。结果表明,采用邻硝基苯甲醛衍生时无法得到相应的衍生化产物。Hunter[13]采用对甲苯亚磺酸钠与ADA在常温下进行反应合成对甲苯磺酰胺基脲,表明对甲苯亚磺酸钠与ADA的反应是非常容易进行的。由于HDC与ADA的化学结构相似,本研究首先采用对甲苯亚磺酸钠对HDC进行衍生化,但仍然无法检测到相应的衍生化产物。目前工业生产ADA发泡剂大多是采用化学氧化剂使HDC氧化为ADA发泡剂,按所用氧化剂的不同又分为铬酸盐法、氯酸盐法、氯气法、过氧化氢法等。但这些方法由于成本高、操作繁琐、生产工艺不易控制等缺点不适合于实验室合成ADA。Bechtold等[12]报道采用高锰酸钾可将HDC氧化为ADA,本研究对该方法进行了一定的改进,采用高锰酸钾对HDC进行氧化,方法简单快速,而且可以在常温下进行。因此,确定的衍生化方法为:先采用高锰酸钾将HDC氧化成为ADA,再采用对甲苯亚磺酸钠对ADA进行衍生化。

表2 不同溶剂对目标物的提取能力(n=2)Table 2 Extractability of different extraction solvents for the analytes(n=2)
本研究以0.06 mol/L高锰酸钾溶液作为氧化剂。首先考察了高锰酸钾溶液用量(80、100、150、200、220、250 μL)的影响。结果表明随着高锰酸钾的用量增加,目标物的峰面积逐渐增大,超过200 μL之后,峰面积变化不大,说明ADA与高锰酸钾已经反应完全。同时考察了氧化时间(10、20、30、40、50、60 min)的影响。当氧化时间为30 min时,目标物的峰面积达到最大,超过30 min,峰面积趋于平衡。因此将氧化条件定为0.06 mol/L高锰酸钾溶液用量为200 μL,氧化时间为30 min。待氧化反应完成后,加入50 μL的二甲基亚砜去除多余的高锰酸钾。
对甲苯亚磺酸钠衍生化的时间与衍生试剂的用量会影响衍生化的效果。通过实验发现ADA与对甲苯亚磺酸钠的反应比较迅速,8 min反应基本完成,10 min以后目标物峰面积基本保持不变,所以本研究确定衍生反应时间为10 min。实验发现,HDC与对甲苯亚磺酸钠的物质的量比在1∶25~1∶30范围内时衍生产物峰面积稳定。对甲苯亚磺酸钠过少可能会使ADA不能快速完全反应,衍生试剂用量过大会造成资源的浪费而且污染环境,综合各种因素选择HDC与对甲苯亚磺酸钠的物质的量比为1∶25,即添加5 mL质量浓度为2.5 mmol/L对甲苯亚磺酸钠溶液进行衍生反应。
2.3 基质效应
采用LC-MS/MS法测定时,普遍存在基质效应,即目标化合物的质谱响应值会受到基质中杂质的影响。本研究采用提取后添加法来评价面粉、馒头、面包、油条、方便面5种基质对目标物响应的影响,通过比较基质匹配曲线和标准品衍生物校正曲线的斜率来考察方法的基质效应(ME),如式(1)所示。当ME<0时,表示目标化合物的MS/MS响应受到抑制;相反,ME>0表示基质增强效应;当ME的绝对值为0~20%表示较弱的基质效应,20%~50%表示中等的基质效应,大于50%表示较强的基质效应[15]。

计算得到5种基质的ME值在-55.8%~-47.3%之间,说明5种基质对目标物的响应值具有较强的抑制效应。当采用外标法进行定量时,HDC的回收率只有48%~71%。为了克服基质效应,往往采用基质匹配溶液的外标法和同位素内标法等方法进行定量[16]。但不同样品的性质差异很大、基质匹配溶液制备工作繁琐等因素限制了基质匹配溶液的外标法的应用。同位素内标与待测物处于同一条件下,受干扰组分的影响几乎一致,可校正目标化合物的响应值,提高方法的精密度和准确度,操作简单;同时同位素内标具有与待测物最为接近的物理化学性质、色谱行为以及响应特征等,在前处理前加入同位素内标可以校正因操作过程造成的待测物的损失[17,18],因此本实验采用同位素内标法定量。当选取稳定同位素标记物13C2,15N2-联二脲作为内标进行定量后,回收率可达到78%~110%,可以看出内标法定量可以使回收率得到明显的改善。
2.4 方法性能
2.4.1 线性范围与检出限
移取适量的标准储备液稀释成质量浓度为1、5、10、100、1 000、20 000 μg/L 的系列标准工作液,按照上述条件进行氧化、衍生与LC-MS/MS分析。以HDC与内标的浓度比为横坐标(x),衍生产物对甲苯磺酰氨基脲的峰面积与13C,15N-对甲苯磺酰氨基脲的峰面积比值为纵坐标(y),进行线性回归分析。结果表明,HDC在1~20 000 μg/L范围具有良好的线性,线性方程为y=1.219 6x+0.030 9,相关系数(R2)为0.999 9。
2.4.2 定量限
分别称取不含HDC的空白基质(面粉、馒头、面包、油条、方便面)2.0 g,分别添加 5 μg/kg的HDC标准品,按照1.2节进行实验与分析,每个样品做6个平行,计算信噪比(S/N)与相对标准偏差(RSDs)。结果表明,HDC的S/N均大于10,且回收率大于70.8%,RSDs小于5.73%。因此将HDC的定量限定为 5 μg/kg。与 Mulder等[10]建立的LC-MS/MS的定量限10 μg/kg进行比较,本研究的定量限低,这表明本方法的灵敏度更高。
2.4.3 回收率和精密度
选取经测定不含有HDC的面粉、馒头、面包、油条、方便面5种基质,分别按照5.0、10.0、50.0 μg/kg 3个浓度水平进行添加回收试验,每个添加水平做6个平行样品,计算平均回收率与精密度,HDC的平均回收率在78.3%~108.0%之间,RSD范围为0.91% ~5.73%(见表3)。与袁丽红等[11]建立的以面粉作为基质的LC-MS/MS方法进行比较,本方法适合于多种面制品基质,实用性更强,更能满足日常检测的需要。

表3 不同面粉制品中HDC的平均回收率和精密度(n=6)Table 3 R esults of recovery test and precision for HDC in fortified flour-based foods(n=6)
2.5 样品测定
ADA加入面粉后,在面团的发酵醒面过程中,开始降解,再经过蒸、炸、煮、烤等加工后,ADA基本上已经降解完全,所以在面制品中基本上没有ADA的存在。应用本方法对市售的馒头、面包等54种面制品中的HDC进行测定,用以监测面粉中是否添加了ADA。结果发现,54个面制品中共有33个样品检出HDC,检出率为61.1%,浓度范围为10.5~29 600 μg/kg,说明这些面粉原料中添加了 ADA。某阳性样品的多反应监测谱图见图4。
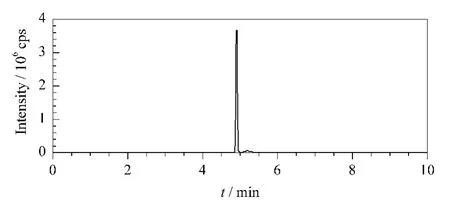
图4 某阳性面包样品的多反应监测图Fig.4 MR M chromatogram of a bread sample
3 结论
本文建立了面粉及其制品中HDC的液相色谱-串联质谱测定方法,采用纯水振荡提取HDC,再使用衍生化法将HDC衍生为对甲苯磺酰氨基脲,通过测定对甲苯磺酰氨基脲来测定HDC的含量,解决了HDC在液相色谱-质谱中响应低的难题。该方法灵敏度高、专属性强,检测范围宽,能满足分析的要求,可作为常规方法对面制品中的HDC进行日常批量分析检测,对面粉中ADA的监管提供了强有力的技术支撑。
[1] Wu Z S,Liang Z Q,Zhong H J,et al.Modern Food Science and Technology(吴正双,梁炽琼,钟海娟,等.现代食品科技),2012,28(9):1239
[2] Long B,Shan J,Yang L J.Food Research and Development(龙彪,单军,杨立军.食品研究与开发),2012,33(7):236
[3] GB2760-2011
[4] Becalski A,Lau B P Y,Lewis D,et al.J Agric Food Chem,2004,52(18):5730
[5] Noonan G O,Begley T H,Diachenko G W.J Agric Food Chem,2008,56(6):2064
[6] Becalski A,Lau B P Y,Lewis D,et al.Food Addit Contam,2006,23(2):107
[7] Noonan G O,Warner C R,Hsu W,et al.J Agric Food Chem,2005,53(12):4680
[8] Zhao X Y,Pan M W,Yuan J F,et al.Journal of Hebei University of Technology(赵晓燕,潘明旺,袁金凤,等.河北工业大学学报),2010,39(3):29
[9] Bechtold W E,Shopp G M,Cheng Y S.J Anal Toxicol,1988,12(2):89
[10] Mulder P P J,Beumer B,Van Rhijn J A.Anal Chim Acta,2007,586(1/2):366
[11] Yuan L H,Ding H L,Chen Y,et al.Science and Technology of Food Industry(袁丽红,丁洪流,陈英,等.食品工业科技),2013,34(10):73
[12] Bechtold W E,Medinsky M A,Cheng Y S,et al.Xenobiotica,1989,19(9):1003
[13] Hunter B A.United States Patent,3903157.1975-09-02
[14] Ye J,Wang X H,Sang Y X,et al.J Agric Food Chem,2011,59(17):9313
[15] Lozano A,Rajski L,Belmonte-Valles N,et al.J Chromatogr A,2012,1268:109
[16] Jia Y B,Wang Q Q,Song H F.Military Medical Sciences(贾彦波,王清清,宋海峰.军事医学),2011,35(2):149
[17] Yan A H,Li X L,Xi C X,et al.Chinese Journal of Analytical Chemistry(严爱花,李贤良,郗存显,等.分析化学),2013,41(4):509
[18] Yang T,Wang J J,Lu Y.Guangzhou Chemical Industry(杨涛,王静静,鹿毅.广州化工),2013,41(12):150
猜你喜欢
中华养生保健(2020年9期)2021-01-18 03:12:36
宜春学院学报(2020年9期)2020-12-03 06:22:10
无机化学学报(2019年2期)2019-02-27 06:53:38
林业与生态(2016年2期)2016-02-27 14:23:55
中国卫生标准管理(2015年6期)2016-01-14 05:17:17
应用化工(2014年7期)2014-08-09 09:20:27
疑难病杂志(2014年12期)2014-04-16 05:19:32
郑州大学学报(工学版)(2014年6期)2014-03-01 04:21:28
哈尔滨医药(2014年5期)2014-02-27 13:35:34
化学分析计量(2013年1期)2013-03-11 16:37:06
