
伴室间隔缺损的Miyoshi肌病的临床、影像和基因一例报道
2014-05-06 06:31:27姚生韩晓琛戚晓昆段枫
中国神经免疫学和神经病学杂志 2014年6期
姚生 韩晓琛 戚晓昆 段枫
伴室间隔缺损的Miyoshi肌病的临床、影像和基因一例报道
姚生 韩晓琛 戚晓昆 段枫
dysferlin蛋白缺陷;临床特点;基因突变
Miyoshi肌病(miyoshi myopathy,MM)和肢带肌营养不良2B型(limb girdle muscular dystrophy type 2B,LGMD2B)的致病基因均为位于染色体2p13的DYSF基因[1]。DYSF基因的cDNA长为69 000,编码产物为dysferlin蛋白[2]。经典的MM主要表现为双下肢远端的肌肉无力和萎缩,后期可以出现双下肢近端的无力,但是伴有先天性室间隔缺损、心脏传导阻滞以及交替性外斜视的MM国内外均未见报道,现报道1例发现Dysferlin蛋白新突变并伴有先天性室间隔缺损、心脏传导阻滞以及交替性外斜视的MM。
1 病例报告 患者,男,21岁,主因“双侧小腿渐进性无力、变细6年”于2011-09-10入院。患者本次入院6年前无明显诱因出现双下肢力弱,活动后易疲劳,渐出现双小腿肌肉萎缩。病情进行性加重,上下楼梯费力,脚尖脚跟走路困难,并出现步态不稳,易跌倒;无肉跳及肌肉酸痛,无头晕头痛及复视,无心悸气短。自发病以来精神、饮食及睡眠正常。既往史:患者于出生后即发现先天性室间隔缺损,1岁时行室间隔缺损修补术;之后多次复查心电图示完全性右束支传导阻滞,心脏超声正常。14岁发现双眼交替性外斜视,注意力集中时眼位可保持正位,后病情进行性加重,双眼斜视幅度加重,于20岁时行双眼外直肌后徙术+内直肌缩短术,术后外斜视消失。足月顺产,产程顺利。否认传染病史,无输血史及药物过敏史;否认毒物、放射物接触史及家族遗传性疾病史。入院后体格检查:生命体征平稳,神志清楚,言语流利,发育正常,营养中等,步入病房,胸骨左缘可见长约5cm的纵行手术疤痕。专科情况:意识清楚,言语流利。高级皮层功能活动正常。右利手。双侧眼球活动自如,无复视,双侧视网膜无色素变性,余脑神经检查未见异常。四肢深浅感觉、关节位置觉正常。双上肢肌力5级;双大腿远端1/3及小腿肌容量减少、肌肉萎缩,无假性肥大;双侧股二头肌、髂腰肌肌力均5级,双侧腹内肌肌力4级,双侧足背屈、趾伸肌力2级。四肢肌张力正常,无震颤及不自主运动。双侧指鼻及轮替试验稳准,双侧跟膝胫试验正常,Romberg征阴性。双侧肱二头肌、肱三头肌腱反射对称引出;双侧膝反射、跟腱反射消失;双侧腹壁反射正常引出;双侧Babinski征、Chaddock征阴性。颈无抵抗,双kernig征及划痕试验阴性。括约肌功能正常。血常规、凝血功能正常;多次检查CK波动在4500~7543 IU/L之间,ALT波动在130~160 IU/L之间;余生化检查正常;乙肝表面抗原、丙肝抗体定性、艾滋病毒抗原/抗体、梅毒血清特异性抗体均阴性,甲状腺功能7项均正常。脑脊液示:脑脊液压力180 mm H2O,脑脊液蛋白125 mg/L、氯化物116 mmol/L、糖2.8 mmol/L,抗神经节苷酯抗体谱均阴性,未见病理性细胞。心电图检查示完全性右束支传导阻滞、右心室高电压。心脏超声检查正常。
对患者行详细的肌电图检查,四肢运动、感觉神经传导均正常,双侧腓肠肌未见运动单位电位;双侧胫前肌大力收缩募集电位呈单纯-混合相。双侧VEP、BAEP、SEP均未见异常。双侧小腿MRI检查示双前后肌群对称性体积变小,肌间隙增宽,肌肉组织内部脂肪变性呈短T1信号(图1 A),脂肪抑制像(STIR)双小腿前后肌群可见到水肿改变(图1B、1C);双侧大腿MRI检查示双侧闭孔外肌、耻骨肌及大腿肌群萎缩脂肪变呈短T1信号(图2A),STIR可见到水肿改变(图2B、2C),尤以大腿远端萎缩为主。右侧胫前肌行肌肉病理检查,肌活检时肉眼观肌纤维完全黄变,光镜下肌纤维脂肪变性呈终末期改变。患者外周血提取DNA后行逆转录合成cDNA,通过PCR技术扩增DYSF基因的编码序列,扩增产物经纯化后序列分析(由北京大学医学部临床遗传系进行基因测定),结果发现DYSF基因第29外显子发现杂合变异位点,为c.3103A>T的错义突变,ACC变为TCC,苏氨酸变为丝氨酸;第54杂合变异位点,为c.6081T缺失的移码突变(图3)。

图1 患者双小腿MRI表现:冠状位扫描示肌群对称性体积变小,肌间隙增宽,肌肉组织内部脂肪变性呈短T1信号(A);STIR示双小腿前后肌群可见到水肿改变(B为冠状位、C为轴位)
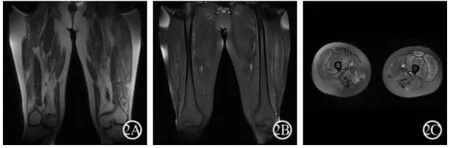
图2 患者双大腿MRI表现:双大腿MRI示双侧闭孔外肌、耻骨肌及大腿肌群萎缩脂肪变呈短T1信号(A);STIR示双大腿肌肉可见到不同程度的水肿改变(B为冠状位、C为轴位)
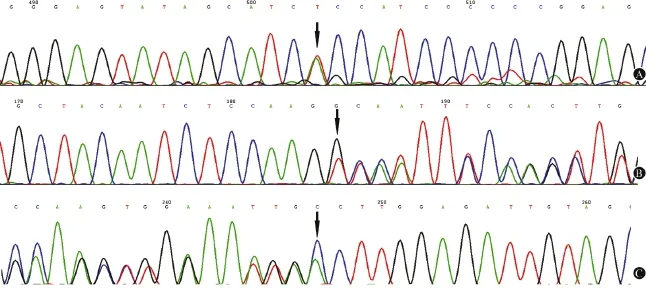
图3 患者外周血提取DNA的DYSF基因检测结果(PCR技术):第29外显子存在杂合变异位点,为c.3103A>T,其所在密码子ACC变为TCC,编码氨基酸由苏氨酸变为丝氨酸,造成错义突变(A);第54外显子序列上存在杂合变异位点,为c.6081T缺失,造成移码突变(B);反向测序进一步证实该缺失突变(C)。图中箭头所示即碱基突变位点
2 讨论 MM是远端性肌病的一种类型,分子生物学研究发现dysferlin蛋白缺陷是该病的致病基因,定位于2p13的DYSF基因。已报道的DYSF基因突变有20多种,突变类型多样,这些突变大多数分布在编码序列的后半段,位于第3外显子到第53外显子之间。骨骼肌定位研究发现dysferlin蛋白主要存在于肌纤维的浆膜上,Dysferlin蛋白主要发挥肌纤维的融合和修复功能[1],当dysferlin蛋白缺乏时,破损的细胞膜不能及时有效修复,继而发生变性坏死。dysferlin蛋白缺陷可以导致LGMD2B和MM两种表型,临床中出现以肩胛带、骨盆带和四肢近端肌无力表现为LGMD2B,而表现为双下肢远端肌无力和萎缩为MM。两种表型之间临床变异较大,甚至在同一种表型之间其临床特点也不完全相同,表现为明显的临床异质性[2]。
该患者青年时渐出现双下肢脚尖、脚跟走路费力,而双上肢活动正常。本次入院后检查发现血CK水平明显增高,波动在4500~7543 U/L之间。双小腿MRI示前后肌群对称性体积变小,肌间隙增宽,肌肉组织内部脂肪变性呈短T1信号,STIR示双小腿前后肌群可见到水肿改变;双大腿MRI示双侧闭孔外肌、耻骨肌及大腿肌群萎缩脂肪变呈短T1信号。STIR示双小腿前后肌群可见到水肿改变,尤以大腿远端肌萎缩为重,提示下肢远端肌肉病变较重[3]。肌电图检查示双侧腓肠肌未见运动单位电位;双侧胫前肌大力收缩募集电位呈单纯-混合相。左侧胫骨前肌活检时肉眼观肌纤维完全黄变,光镜观察肌纤维完全脂肪纤维化呈终末期改变。患者外周血DNA检查发现DYSF基因第29外显子出现杂合变异位点,为c.3103A>T的错义突变,苏氨酸变为丝氨酸(ACC变为TCC);第54外显子为杂合变异位点,为c.6081T缺失的移码突变。患者的临床、MRI、病理改变以及DYSF基因突变特点支持MM的诊断。但该患者伴有的先天性室间隔缺损、完全性右束支传导阻滞以及交替性外斜视国内外均未见报道[4]。
该患者除了MM比较典型的临床表现之外,还伴有先天性室间隔缺损、心脏传导阻滞以及青少年时期出现的双眼交替性外斜视。由于本例患者dysferlin蛋白突变为一新发突变,由于突变位点不同,其临床表现各异,或许可以解释患者除骨骼肌累及以外,心肌以及眼外肌也可累及的特点。该患者出生后发现胸憋及心功能不全,心脏超声发现先天性室间隔缺损,经室间隔缺损修补术后患者心功能恢复正常,后多次复查心电图发现完全性右束支传导阻滞。研究发现dysferlin蛋白在多种脏器均可以表达,尤以骨骼肌、心肌和肾脏表达明显增高[5]。dysferlin蛋白缺陷时高表达的器官和组织如骨骼肌最易累及且病情较重,而心脏和肾脏累及报道较少。Wenzel等[6]报道了7例dysferlinopathy(即LGMD2B),其中2例患者出现进行性的呼吸困难,检查发现为扩张型心肌病,其他5例患者尽管没有心脏方面的症状,但检查发现左室肥大和心肌复极异常。Choi等[7]通过2维超声和心肌MRI增强扫描对MM患者的心脏进行检查,发现MM患者心肌出现临床下损害,随着病情进展,可能会发展为心肌病而出现心脏方面的表现。这些研究提示dysferlin蛋白缺陷可以导致心肌受累而表现为心肌损害以及心律失常。该患者在14岁并发双眼交替性外斜视,是否也与dysferlin蛋白缺陷有关值得探讨。导致双眼交替性外斜视的病因较多,儿童发病多见,多与先天发育异常、疲劳、遗传等因素有关。该患者家族中未发现有交替性外斜视的病例,且在国内外已报道的MM患者中未报道伴有先天性室间隔缺损以及交替性外斜视的患者,是否也与该患者的dysferlin蛋白突变位点相关,也需要进一步证实。
参考文献:
[1]Kerr JP,Ziman AP,Mueller AL,et al.Dysferlin stabilizes stress-induced Ca2+signaling in the transverse tubule membrane[J].Proc Natl Acad Sci U S A,2013,110(51):20831-20836.
[2]孙顺昌,樊绮诗,吴华成,等.Dysferlin缺陷:肢带2B型肌营养不良与Miyoshi肌病的致病原因[J].中华医学遗传学杂志,2004,21(2):128-131.
[3]Kesper K,Kornblum C,Reimann J,et al.Pattern of skeletal muscle involvement in primary dysferlinopathies:a wholebody 3.0-T magnetic resonance imaging study[J].Acta Neurol Scand,2009,120(2):111-118.
[4]Zhao Z,Hu J,Sakiyama Y,et al.DYSF mutation analysis in a group of Chinese patients with dysferlinopathy[J].Clin Neurol Neurosurg,2013,115(8):1234-1237.
[5]Chase TH,Cox GA,Burzenski L,et al.Dysferlin deficiency and the development of cardiomyopathy in a mouse model of limb-girdle muscular dystrophy 2B[J].Am J Pathol,2009,175(6):2299-2308.
[6]Wenzel K,Geier C,Qadri F,et al.Dysfunction of dysferlindeficient hearts[J].J Mol Med,2007,85:1203-1214.
[7]Choi ER,Park SJ,Choe YH,et al.Early detection of cardiac involvement in Miyoshi myopathy:2D strain echocardiography and late gadolinium enhancement cardiovascular magnetic resonance[J].J Cardiovasc Magn Reson,2010,12(1):31.
R746.9
:D
:1006-2963(2014)06-0449-03
2014-06-05)
(本文编辑:邹晨双)
10.3969/j.issn.1006-2963.2014.06.021
100048北京海军总医院神经内科
戚晓昆,Email:bjqxk@sina.com
猜你喜欢
宁夏医学杂志(2020年3期)2021-01-21 08:23:42
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:58
天津医科大学学报(2019年6期)2019-08-13 07:04:34
中国卫生标准管理(2015年8期)2016-01-15 03:58:44
少年科学(2015年4期)2015-05-07 04:03:25
中国当代医药(2015年30期)2015-03-01 02:08:07
中国当代医药(2015年29期)2015-03-01 02:07:48
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:55
雷达学报(2014年4期)2014-04-23 07:43:09
湖南中医药大学学报(2013年5期)2013-03-11 16:33:35
