
南海南部海域浮游细菌群落特征及影响因素研究
2014-04-28 03:58:50刘小沙赵阳国高会旺中国海洋大学环境科学与工程学院海洋环境与生态教育部重点实验室山东青岛266100
中国环境科学 2014年11期
白 洁,刘小沙,侯 瑞,赵阳国,高会旺 (中国海洋大学环境科学与工程学院,海洋环境与生态教育部重点实验室,山东 青岛 266100)
南海南部海域浮游细菌群落特征及影响因素研究
白 洁,刘小沙,侯 瑞,赵阳国*,高会旺 (中国海洋大学环境科学与工程学院,海洋环境与生态教育部重点实验室,山东 青岛 266100)
采用高通量测序技术,对南海南部海域浮游细菌丰度、群落组成和群落多样性的分布特征及与环境因子的关系进行了研究.结果表明,该研究区域浮游细菌丰度为107~108个/L,近岸大于离岸,同一站位不同水层细菌分布差异明显.优势类群为变形菌门、蓝藻门和拟杆菌门,优势亚群为 γ-变形菌纲、α-变形菌纲、蓝藻菌纲和黄杆菌纲,研究区域内不同水体间物种组成存在较大差异,另外该海域还存在大量未被认知的细菌类群.该海域浮游细菌种类丰富,具有较高的多样性指数(H′)(4.44~7.00),研究区域内表层水体H′接近,分别为5.26、5.33和5.07,处于上升流的次表层水体中H′为6.70明显高于其他水层.DOC和磷酸盐是影响该海域浮游细菌丰度的主要因素,同时磷酸盐也是影响其群落多样性的主要因素,表明该海域异养浮游细菌生长主要受P的限制.
南海南部;浮游细菌;16S rDNA;群落结构;环境因子
海洋浮游细菌是海洋生态系统的重要组成部分,参与物质循环和能量流动,在海洋生态系统中发挥着重要的生态作用.海洋浮游细菌种类丰富、数量庞大,不同微生物群落具有其独特的生态功能,构成了微生物功能多样性.
随着分子生物学的发展,DNA测序技术的广泛应用,以环境DNA为模板进行微生物群落组成分析已成为环境微生物多样性研究的一种有效手段[1].高通量测序技术以低成本、高通量、流程自动化为优势,广泛应用于基因组测序和表观基因组学以及功能基因组学研究的许多方面[2],也迅速发展为微生物群落结构研究中最先进的方法之一.
南海位于西太平洋,是我国面积最大的海域,南侧延至赤道,终年高温,受季风、地形及黑潮等影响,水文条件较复杂,独特的地理环境使其具有丰富的微生物资源.对我国南海微生物多样性的研究多集中在南海中部、北部海区[3]以及南沙海域的沉积物[4].南海南部海域是连结热带西太平洋和印度洋的热带边缘海,地理环境较为复杂,对此海域浮游细菌多样性的研究鲜有报道.
本研究选取了南海南部海域3个典型站位,采用高通量测序技术对浮游细菌群落16S rDNA多样性进行分析,研究了其群落组成和分布特征,并进一步探讨了细菌丰度分布、群落结构与环境因子间的关系.
1 材料与方法
1.1 样品采集、处理与现场环境参数测定
于2012年8月在南海南部海域设定3个典型站位进行样品采集和现场观测,S1和S3站基本处于同一纬度,其中S1站离岸相对较远,S3站离岸相对较近;S2站距赤道较近,站位布设见图1.
图1 采样站位示意Fig.1 Sampling stations
海水样品由船载CTD单次采集,其中S1和S2站采集表层(3m)和次表层(75m)海水, S3站仅采集表层水(3m).采集的部分水样加入无颗粒多聚甲醛固定,4℃保存,用于细菌数量测定;部分水样2L经0.22μm滤膜过滤,-80℃保存滤膜,用于细菌群落结构分析;部分水样过滤后4℃保存,用于DOC和营养盐分析.现场海水温度、pH值、盐度和溶解氧等环境参数由CTD(SBE9 型)直接测得.细菌丰度和理化性质测定,均设定3个平行样.
1.2 DOC和营养盐测定
DOC测定采用高温催化氧化法,由岛津TOC-Vcpn总有机碳分析仪测定.营养盐由BRAN+LUEBBE AA3型营养盐自动分析系统进行测定,其中NO3--N采用Cu-Cd还原法,NO2--N采用盐酸萘乙二胺络合显色法,NH4+-N采用靛酚蓝法,PO43-采用磷钼蓝法.
1.3 细菌丰度测定
浮游细菌数量采用荧光显微镜计数法进行测定,样品经DAPI染色,采用LeicaDMLA型全自动落射荧光显微计数.
1.4 高通量测序及分析
采用Power Soil DNA提取试剂盒(Mobio, USA)提取滤膜上总DNA,最终溶解于2mmol/L Tris-HCl (pH=8)中,取5µL DNA进行电泳检测.
以总DNA为模板,以细菌16S rDNA的V1~V3区为目标,采用融合了454测序平台的通用引物进行PCR扩增.融合V1~V3区的正向引物:CGTATCGCCTCCCTCGCGCCATCAG+BAC ODE+PRIME F;融合V1~V3区的反向引物:CTATGCGCCTTGCCAGCCCGCTCAG+PRI ME R).PCR体系为50µL,反应条件为:94 ℃预变性30s, 94℃ 20s, 45℃ 20s, 65℃ 60s,循环5次;94℃ 20s, 60℃ 20s, 72℃ 20s,循环20次;72℃延伸5min.PCR结束后,对PCR产物进行琼脂糖电泳,采用琼脂糖回收试剂盒(上海生工)对DNA进行回收.
PCR回收产物用Qubit2.0定量,根据测得的DNA浓度,将所有样品按照1:1的比例进行混合;混合后充分震荡均匀,混合产物应用454测序平台测序,测序委托上海生工进行.对原始序列进行预处理,选取高质量的序列进行分析,获得的序列平均长度在450bp以上.
采用RDP classifier 软件对预处理序列进行物种分类,对每条序列在genus水平上计算其分配到此rank中的概率值,大于0.8则说明此分类结果可信.
OTU聚类采用uclust软件,首先筛选出序列中最长的reads作为种子序列,找出所有与该序列相似度在阈值范围内(>97%)的序列,并将其归为一个类,依次重复此步骤,直到所有序列均聚好类,每一个类作为一个OTU[5].
1.5 细菌群落多样性
Alpha多样性采用香农指数(H')进行分析.香农指数(H')衡量群落的异质性,计算用式(1):

其中:Pi为各种群物种数与样本总物种比值,并以从样本中随机抽取到的序列数为横坐标绘制稀疏分析图.
Beta多样性采用样本PCoA分析法,此法基于Unifrac metric[6]来衡量样本间物种组成的相似度,本研究采用加权重计算方式计算Unifrac值.加权重的计算方式计算时不仅评估样本间物种的差异,并且加入了物种丰度(即OTU分配到的reads数及样本总reads)作为权重.
群落多样性、细菌丰度与环境因子之间的相关性采用SPSS19.0软件进行分析.
2 结果
2.1 浮游细菌丰度的分布特征
本研究海域3个站位浮游细菌丰度分布见图2,其平均值范围为107~108个/L,与Chen等[7]在南海南部测定的浮游细菌丰度接近,但小于Ning等[8]测定的南海北部细菌丰度108~1010个/L.由图2可见,离岸较近的S3站表层水体含有细菌数量最多,与之相比,处于同一纬度离岸较远的S1站细菌数量较小,靠近赤道的S2站的表层含有的细菌数量最少.在垂直方向上细菌丰度分布差异更加明显,S1站的表层细菌丰度大于其次表层,而S2站次表层的细菌丰度明显大于其表层水体.细菌丰度分布的空间差异可能是营养物质含量及水文条件等环境因子共同影响的结果.

图2 3个站位的细菌丰度分布Fig.2 Bacterial abundance in three stations
2.2 16S rDNA序列分析及群落特征
采用高通量测序法对南海南部海域3个站位不同水层的5个样品所提取的16S rDNA基因片段进行序列分析,共获得66118条有效序列和9316个OTU,其中S2站次表层水体(S2b)的OTU最多,为6775个;S3站表层水体(S3s)的最少,为889个;各样品文库的覆盖率为84%~94%(表1).通过RDP classifier程序对不同样品的基因序列从门到属依次进行分类,结果见表1.
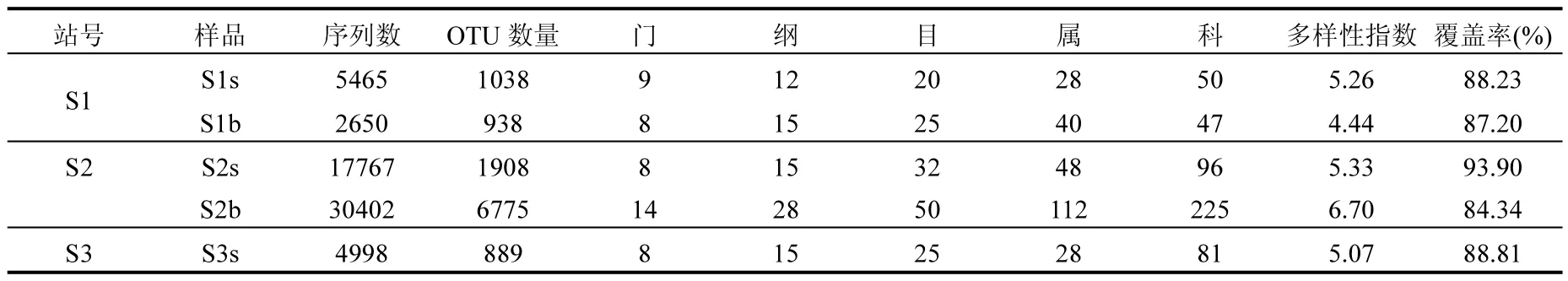
表1 细菌16S DNA序列分析Table 1 Sequential analysis of bacterial 16S DNA
本研究海域共获得浮游细菌14门、28纲、54目、126属、281科,主要类群组成见图3.
在门水平上,S2站的次表层水样S2b包含了所有的门类,S1b、S2s、S3s分别包含8门,S1s含有9门.其中S1s、S2s、S2b、S3s的优势菌群均为变形菌门(Proteobacteria),分别占其总群落的70.01%、62.85%、81.47%、58.14%,第二优势菌群为蓝藻门(Cyanobacteria).S1b的优势菌群蓝藻门(Cyanobacteria)所占比例大于变形菌门(Proteobacteria),分别为41.47%和40.23%.另外,拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)和浮霉菌门(Planctomycetes)在所有水样中均存在.在分类结果中有5门仅存在S2b中,所占比例均小于0.2%,分别为绿弯菌门(Chloroflexi)、黏胶球形菌门(Lentisphaerae)、侯选门OP10(Armatimonadetes )[9-10]、酸杆菌门(Acidobacteria)和绿菌门(Chlorobi).
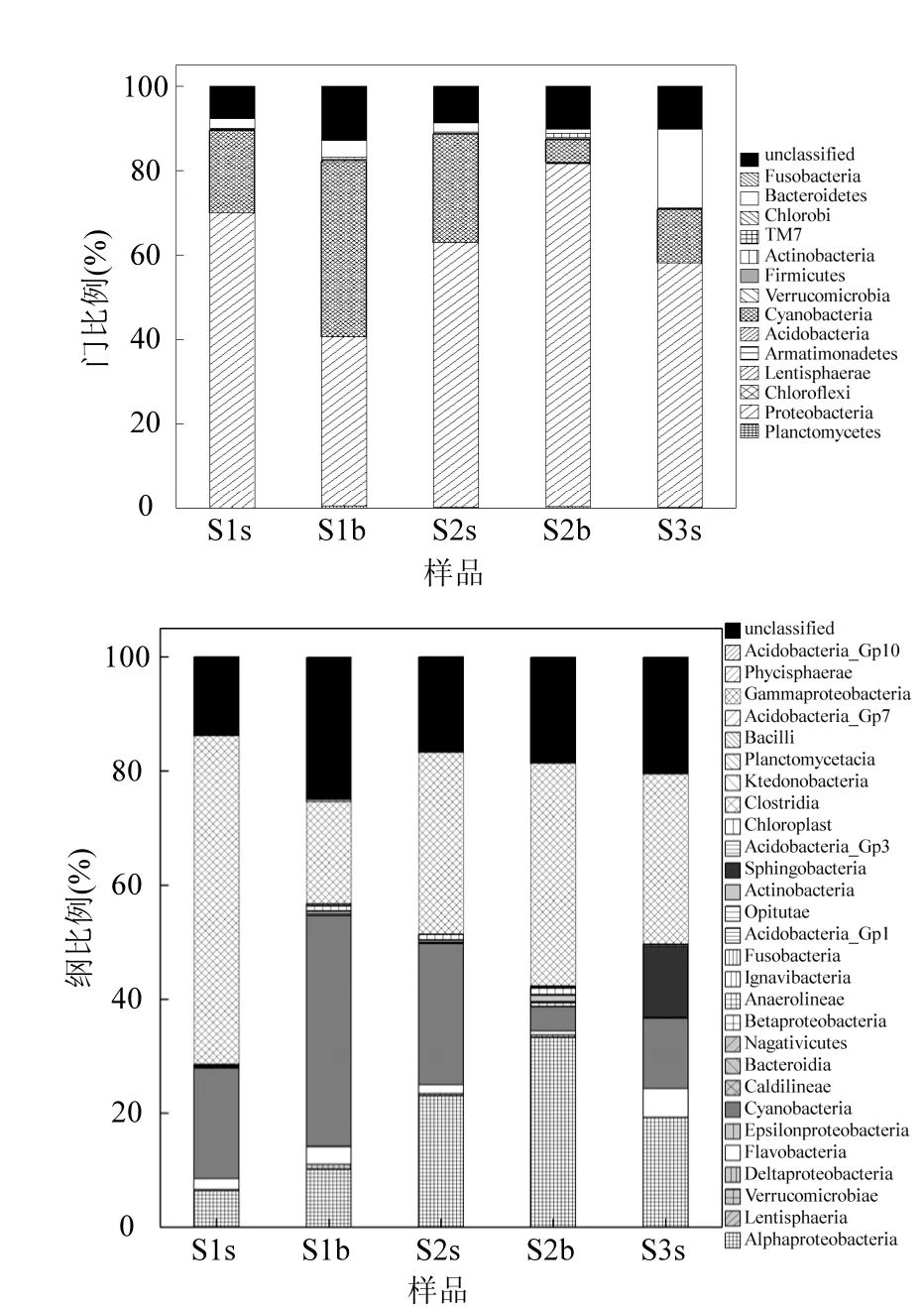
图3 不同水样的群落组成Fig.3 The bacterial community composition in different water samples
在纲水平上,S1s包含12纲,S1b、S2s、S3s均包含15纲,而S2b包含了28纲.S1s、S1b、S2s以及S2b的优势菌群较为相似,均包含 γ-变形菌纲、α-变形菌纲、蓝藻菌纲、黄杆菌纲,S3s优势菌群除这4纲外还包含了鞘脂杆菌纲(12.4%);S1b最大的优势菌群为蓝藻菌纲,其他水样均为 γ-变形菌纲.另外,由图3可见,S1和S2站次表层水样(S1b和S2b)中α-变形菌所占比例均大于其表层水样(S1s和S2s).
此外,每个水样中都含有大量无法确定其分类位置的序列,表明该海域存在大量未被认知的细菌类群.
2.3 浮游细菌多样性和群落组成差异分析
通过计算每个样品的香农指数(H′)来衡量各个水样群落的异质性,本研究海域浮游细菌生物多样性分布见图4,可以看出各站位不同水层浮游细菌的香农指数(H′)均相对较高,但存在较大差异,表明该海域浮游细菌生物多样性较高.3个表层水体S1s、S2s和S3s的H′较为接近,分别为5.26、5.33和5.07;而S1和S2站次表层水体的H′值差异较大,其中S1b的H′是4.44,S2b的H′是6.70.
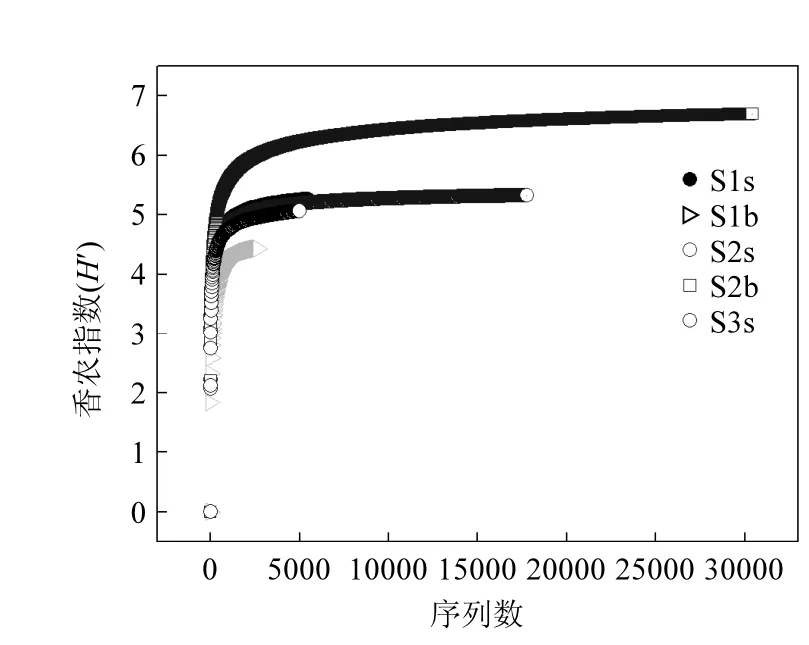
图4 不同水样的香农指数(H′)Fig.4 The Shannon index in different stations
采用PCoA分析衡量样本间物种组成的相似度,用加权重(weighted)计算方式获得的Unifrac值见图5.从图5可以看出,不同水体间物种组成存在较大差异,S1的表层(S1s)和次表层(S1b)距离最近,物种组成最相似;S2表层(S2s)和S3站表层(S3s)距离相近,物种组成较相似;而S2b与其他水体的距离都很远,说明S2b与其他区域的物种组成存在较大差异.
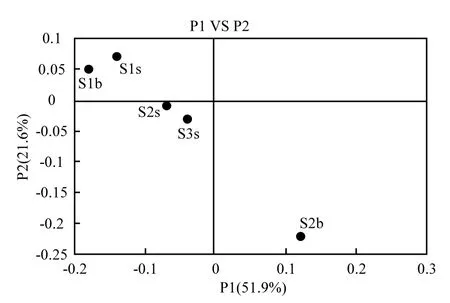
图5 不同水样物种组成相似度分析(weighted PCoA)Fig.5 The similarity analysis of species composition among different stations
3 讨论
本研究通过对南海南部海域3个站位共5个水样进行分析,表明该海域浮游细菌丰度分布存在明显的空间差异;不同水样中获得的细菌基因序列数和OTU差异较大,并存在共同的优势菌群,但群落多样性各不相同,说明该研究海域微生物种类丰富,群落组成复杂,海洋环境的不同可能是导致微生物多样性的重要原因.
3.1 浮游细菌类群与生态功能
本研究海域5个水样测得的细菌种类丰富,不同水样中细菌群落组成具有相似性.5个水样中变形菌门(Proteobacteria)为共同的优势类群,在各个水样中所占比例在40%~70%之间,这一结果与国内外海洋微生物多样性研究[11-15]结果一致.
本研究优势亚群为γ-和α-变形菌纲,其中γ-变形菌纲比例明显大于α-变形菌纲.大量西太平洋海域研究表明γ-变形菌纲为其优势菌群[16-18],在波罗的海研究中发现在海水中γ-变形菌纲含量最大[19].γ-变形菌纲是目前所知的细菌中种类最多的一纲,包括一些重要的类群,其中交替单胞菌属为化能异养菌,分子氧是一般的电子受体,部分能够进行反硝化,海洋中营养物质和含氧量可能会影响其分布.Jing等[15]发现在南海深水层中存在大量的假交替单胞菌属,其细菌能分泌多种胞外活性物质,有助于获取营养和竞争生存空间等,具有重要的生态学作用.
本研究中α-变形菌纲为变形菌门(Proteobacteria)中第二大优势菌群,主要包含根瘤菌目、鞘脂单胞菌目和红杆菌目,其中红杆菌目微生物在S2站的次表层水体S2b中所占比例最大,为22.27%.Giovannoni等[20]发现红杆菌目和SAR11占表层海水微生物的30~50%;Jing等[15]在研究热带和亚北极细菌群落分布中发现在南海海域、哥斯达黎加圆突区和亚北极海域中上部水层中(表层和次表层)α-变形菌纲所占比例最大.α-变形菌纲中的红杆菌目细菌为光合异养菌,能够利用环境中的硫化物和有机底物,根瘤菌目中大部分细菌可以进行固氮,因此这些微生物在海洋的碳、氮、硫循环中起着非常重要的作用.
蓝藻门(Cyanobacteria)为本研究大部分水体的第二优势菌群,优势亚门为蓝藻菌纲,主要的蓝细菌为GpIIa,在其他海域的研究[21-22]中也有类似结论.本研究蓝藻门在S1站次表层(S1b)分布最多,S2站的次表层(S2b)分布最少,各水样中所占比例为5%~42%.蓝藻门(Cyanobacteria)细菌为产氧光合细菌,是海洋生态系统和初级生产力的重要组成部分,同时在海洋碳、氮、硫的循环中发挥重要作用.
拟杆菌门(Bacteroidetes)为5个水样的第三大优势菌群,主要包括黄杆菌纲和鞘脂菌纲,前者丰度大于后者,在波罗的海研究中也发现类似的结果[19],本研究黄杆菌纲主要分布在表层海水中(S1s和S3s),因其能够产生胞外水解酶来降解大分子有机物质,是所在水域碳循环中的重要功能类群.
另外,本研究所有水样中均发现放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)和浮霉菌门(Planctomycetes)的存在,其比例均小于0.3%.厚壁菌门多为革兰氏染色阳性,在近海和浅海沉积物中较为常见[23], Jing等[15]在南海海水中也发现了放线菌门、疣微菌门和浮霉菌门,但未发现本研究各水样中均存在的厚壁菌门.
3.2 浮游细菌丰度与环境因子的关系
3个站位浮游细菌丰度与主要环境因子之间的关系见表2,研究结果发现,细菌丰度与DOC(P≤0.01)、磷酸盐(P≤0.05)均呈显著性正相关关系,与其他环境因子之间的相关性不显著.
在海洋生态系统中,DOC是细菌维持生命活动、进行生长繁殖的营养来源,对细菌生产力起着决定性的作用.早在20世纪末Rivkin等[24]和Pomeroy等[25]就提出,细菌生长受海水温度、DOC和无机营养盐的限制.Hitchcock等[26]的实验表明,向模拟环境中添加一定浓度的DOC,浮游细菌数量在短时间内明显增加.本研究S1和S3站处于同一纬度,但S3站离岸较近, DOC含量丰富,细菌数量大,而S1离岸较远,DOC含量很低,细菌数量较小,推测可能是含有丰富有机质的陆源水体进入S3站附近海域,促进了临近水域浮游细菌的生长繁殖.
海洋中异养浮游细菌的生长还依赖于无机营养盐的种类和数量,异养浮游细菌可以利用海水中的溶解性磷酸盐作为P源进行生长繁殖.Cotner等[27]通过海洋调查发现,Sargasso Sea海区DOC含量很高但细菌生产力较低,向实验水体添加无机磷酸盐后,异养浮游细菌的生物量和生产力均明显增加,认为正是由于磷的限制,才导致细菌二次生产力的下降.南海为寡营养盐海区,本研究海域中除S2b和S3s外磷酸盐含量均较低,结合细菌丰度与磷酸盐存在显著性正相关的结果可以初步推断,南海南部海域异养浮游细菌生长受P的限制.

表2 不同站位主要环境因子及与细菌丰度和群落多样性之间的关系Table 2 The main environmental factors and their correlation with bacterial abundance and biodiversity
3.3 浮游细菌类群分布与环境因子的关系
由表2可见,研究海域细菌生物多样性与磷酸盐含量之间存在显著性正相关关系(r=0.836 P≤0.05),而与其他环境因子之间的相关性不显著.刘敏[28]在研究我国黄、东海典型生态过程中的微生物群落结构也发现磷酸盐是控制群落分布的主要因子,另外Haukka等[29]也报道在淡水生态系统中添加磷酸盐和总氮后,浮游细菌的群落结构发生变化,这种变化可能与生态系统中添加营养物质后引起的水体富营养化有关.
本研究中S2b的H′明显高于其他区域,可能与该区域的化学、水文条件等多种因素有关.S2b位于S2站的次表层,水温为23.7℃,明显低于同纬度其他水域;其盐度为33.9,DIN为8.65μmol/L,磷酸盐为0.25μmol/L(见表2),均为5个水体中的最高值, DOC含量也仅低于S3s,推断S2站可能处于上升流区.底层海水上升带入丰富的营养盐,有利于浮游藻类的生长繁殖,并造成海水DOC的增加,从而导致细菌的旺盛生长,使细菌丰度明显增加,群落结构更加复杂.本研究中S2b包含的细菌门类最多,绿弯菌门(Chloroflexi)、黏胶球形菌门(Lentisphaerae)、侯选门OP10 (Armatimonadetes)、酸杆菌门(Acidobacteria)和绿菌门(Chlorobi)仅存在于该区域的结果也与其特殊的理化条件有关.有研究表明,α-变形菌广泛存在于富营养化水体中,本研究S2b中α-变形菌数量最多,应该与其营养物质含量高有关.
此外,Agogu等[30]指出海水中细菌的组成由于潮流的影响会迅速发生变化,因此,细菌群落组成与多样性的空间差异受到多种因素的共同影响,评价微生物群落与环境因素之间的关系进行多种因素的综合影响分析更为科学.
4 结论
4.1 南海南部海水中细菌群落组成丰富,优势类群为变形菌门(Proteobacteria)、蓝藻门(Cyanobacteria)和拟杆菌门(Bacteroidetes);另外拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)、疣微菌门(Verrucomicrobia)和浮霉菌门(Planctomycetes)在所有水样中均存在.优势亚群为 γ-变形菌纲、α-变形菌纲、蓝藻菌纲、黄杆菌纲.另外每个水样中均含有大量无法确定其分类位置的序列,表明该海域存在大量未被认知的细菌类群.
4.2 细菌的丰度和群落组成存在明显的空间差异.细菌丰度分布近岸大于离岸,垂直分布规律不明显.S1站的表层和次表层群落组成相似,S2站的表层与S3站的表层群落组成相似.研究区域群落多样性指数H′较高,表层水体H′相近;与其他站位相比,处于上升流的S2站的次表层的群落组成更加复杂,H′明显偏高.
4.3 本研究海域发现细菌丰度与DOC、磷酸盐呈显著正相关关系,表明DOC和磷酸盐是影响该海域浮游细菌丰度的主要因素,而细菌群落多样性仅与磷酸盐存在显著正相关关系,表明该海域异养浮游细菌生长受P的限制;该海域群落组成与多样性的空间差异可能是水文条件,地理位置等多种因素综合影响的结果.
[1] Nocker A, Burr M, Camper A K. Genotypic microbial community profiling: a critical technical review [J]. Microb. Ecol., 2007,54: 276-289.
[2] Mardis E R. Next-generation DNA sequencing methods [J]. Annual Review Genomics and Human Genetics, 2008,9:387-402.
[3] 周宗澄,姚瑞梅,梁子原.南海中部海域异养细菌的生态分布 [J].海洋通报, 1989,8(3):57-64.
[4] 戴 欣,周 慧,陈月琴.中国南海南沙海区沉积物中细菌16s rDNA多样性的初步研究研究 [J]. 自然科学进展, 2002,12(5): 479-484.
[5] Cole J R, Chai B. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis [J]. Nucleic Acids Research, 2005,33:294-296.
[6] Lozupone C, Knight R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities [J]. Applied and environmental microbiology, 2005,71(12):8228-8235.
[7] Chen B Z, Liu H B, Wang Z L. Trophic interactions within the microbial food web in the South China Sea revealed by size-fractionation method [J]. Experimental Marine Biology and Ecology, 2009,36(8):59-66.
[8] Ning Xiuren, Li William K W, Cai Yuming. Comparative analysis of bacterioplankton and phytoplankton in three ecological provinces of the northern South China Sea [J]. Marine Ecology Progress Series, 2005,293:17-28.
[9] Thompson F L., Bruce T. Coastal bacterioplankton community diversity along a latitudinal gradient in Latin America by means of V6tag pyrosequencing [J]. Arch Microbiol, 2011,193:105-114.
[10] Portillo M C, Gonzalez J M. Members of the candidate division OP10are spread in a variety of environments [J]. World Journal of Microbiology and Biotechnology, 2009,25(2):347-353.
[11] 卢婧雯,张心齐,杜丽丽.中国东海及南海近海4采样点海水可培养细菌的多样性研究 [J]. 浙江大学学报, 2012,39(4):443-449.
[12] 白 洁,李海艳,赵阳国.黄海北部不同站位海洋细菌群落分布特征 [J]. 微生物学报, 2009,49:343-350.
[13] Zeng Y X, Zhang F, He J F. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes [J]. Antonie Van Leeuwenhoek, 2013,103:1309-1319.
[14] Siam R, Mustafa G A, Sharaf H, et al. Unique prokaryotic consortia in geochemically distinct sediments from Red Sea Atlantis II and discovery deep brine pools [J]. PLoS ONE, 2012,7(8):112-121.
[15] Jing H, Xia X, Suzuki K, Liu H. Vertical profiles of bacteria in the tropical and subarctic oceans revealed by pyrosequencing [J]. PLoS ONE, 2013,8(11):321-327.
[16] 谢 华,薛燕芬,赵爱民.太平洋帕里西维拉海盆细菌多样性的非培养的初步分析 [J]. 微生物学报, 2005,45(1):1-5.
[17] 李会荣,俞 勇,曾胤新.北极太平洋扇区海洋沉积物细菌多样性的系统发育分析 [J]. 微生物学报, 2006,46(2):177-183.
[18] Newberry C J, Webster G, Cragg B. Diversity of prokaryotes and methanogenesis in deep subsurface sediments from the Nankai Trough, Ocean Drilling Program Leg190 [J]. Environ. Microbiol., 2004,6(3):274-287.
[19] Koskinen K, Hultman J, Paulin L, et al Spatially differingbacterial communities in water columns of the northern Baltic Sea [J]. FEMS Microbiol Ecol, 2011,75:99-100.
[20] Giovannoni S J, Tripp H J, Givan S, et al. Genome streamlining in a cosmopolitan oceanic bacte-rium [J]. Science, 2005,309: 1242-1245.
[21] Jing H M, Liu H B. Phylogenetic composition of Prochlorococcus and Synechococcus in cold eddies of the South China Sea [J]. Aquat Microb Ecol, 2012,65:207-219.
[22] Flombaum P, Gallegos J L, Gordillo R A, et al. Present and future global distributions of the marine Cyanobacteria Prochlorococcus and Synechococcus [J]. PNAS, 2013,110(64):9824-9829.
[23] Ravenschlag K, SahmK, Peruthaler J. High bacterial diversity in permanently cold marine sediments [J]. Applied and Environmental Microbiology, 1999,65:3982-39891.
[24] Rivkin R B, Anderson M R. Inorganic nutrient limitatio n of oceanic bacter-ioplankton [J]. Limnology and Oceanography, 1997,42:730-740.
[25] Pomeroy L R, Deibel D. Temperature regulation of bacterial activity during the spring bloom in New found land coastal waters [J]. Science, 1996,233:359–361.
[26] Hitchcock J N, Mitrovic S M. Responses of estuarine bacterioplankton, phytoplankton and zooplankton to Dissolved Organic Carbon (DOC) and Inorganic Nutrient Additions [J].Estuaries and Coasts, 2010,33:78-91.
[27] Cotner J B, Ammerman J W, Peele E R. Phosphorus limited bacterioplankton growth in the Sargasso Sea [J]. Aquatic Microbial Ecology, 1997,13:141-149.
[28] 刘 敏.我国黄、东海典型生态过程中微生物群落结构 [D]. 青岛:中国科学院海洋研究所, 2011.
[29] Haukka K, Kolmonen E, Hyder R, et al. Effect of nutrient loading on bacterioplankton community composition in lake mesocosms [J]. Microb. Ecol, 2006,51:137-146.
[30] Agogue H, Gasamayor E O, Bourrain M. A survey on bacteria inhabiting the sea surface microlayer of coastal ecosystems [J]. FEMS Microbiology Ecology, 2005,54:269-280.
Community structure and influencing factors of bacterioplankton in the southern South China Sea.
BAI Jie, LIU Xiao-sha, HOU Rui, ZHAO Yang-guo*, GAO Hui-wang (Key Laboratory of Marine Environmental Science and Ecology, Ministry of Education, Ocean University of China, Qingdao 266100, China). China Envrionmental Scicence, 2014,34(11):2950~2957
The high-throughput sequencing approach was adopted to analyze the distribution characteristics of the abundance, community structure and community diversity of bacterioplankton in the southern South China Sea and their relationship with environmental factors. The results indicated that the bacterioplankton abundance in the investigated area was about 107~108cells/L with high values in offshore stations and sharp differences among water layers. The dominant groups were Proteobacteria, Cyanobacteria and Bacteroidetes and the dominant classes were γ-Proteobacteria, α-Proteobacteria, Cyanobacterium and Flavobacteria. The community structure of bacterioplankton showed an obvious difference among various water bodies and numerous unidentified bacteria were recorded. The bacterioplankton showed a high species richness in the area with a biodiversity index (H') of between 4.44~7.00. The biodiversity index in surface layers of the area was 5.26, 5.33 and 5.07, respectively, whereas the index in the upwelling subsurface layer was 6.70, which was significantly higher than other layers. DOC and phosphate were the main factors influencing the bacterioplankton abundance, phosphate was also the main factor affecting the community diversity. The growth of bacterioplankton in the southern South China Sea was therefore mainly limited by phosphate.
southern South China Sea;bacterioplankton;16S rDNA;community structure;environmental factor
X171.1,Q938
A
1000-6923(2014)11-2950-08
白 洁(1962-),女,陕西神木人,教授,博士,主要从事海洋微生物生态学研究.发表论文30余篇.
《中国环境科学》获评“百种中国杰出学术期刊”
《中国环境科学》编辑部
2014-02-28
国家自然科学基金重大国际合作研究项目(41210008)
* 责任作者, 副教授, ygzhao@ouc.edu.cn
《中国环境科学》2012年被中国科学技术信息研究所评为“2011年度百种中国杰出学术期刊”.“百种中国杰出学术期刊”是根据中国科技学术期刊综合评价指标体系进行评定的,包含总被引频次、影响因子、基金论文比、他引总引比等多个文献计量学指标.
猜你喜欢
海洋石油(2021年3期)2021-11-05 07:43:10
河北环境工程学院学报(2021年1期)2021-03-19 08:43:00
潍坊学院学报(2020年2期)2021-01-18 07:02:00
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
流行色(2019年10期)2019-12-06 08:13:26
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
环境科技(2016年2期)2016-11-08 12:18:22
焊接(2015年6期)2015-07-18 11:02:25
