
香豆素衍生物与人血清白蛋白的相互作用
2014-04-27 08:44朱晋娴
科技视界 2014年8期
李 凌 朱晋娴
(徐州医学院 麻醉学院,江苏 徐州 221004)
通过实验表征,研究药物的作用机理,实际上就是研究药物与靶点的相互作用,结合计算化学,分别成为药物分子设计的实验和理论基础。人血清白蛋白HSA是血浆中含量最高的蛋白质,是不同组织氨基酸的主要来源,又是大量内源性物质如长链脂肪酸﹑胆红素及外源性物质如药物在体内的转运蛋白,它可以携带药物至作用靶点,进而发挥药物的活性。药物与白蛋白的不同键合会影响到药物的治疗作用﹑药代动力学及其毒性,同时也对药物在体内的分布起着重要的作用。
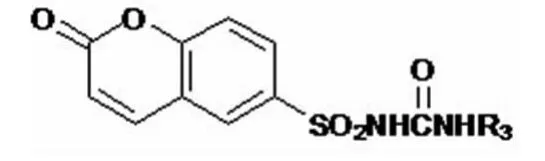
以香豆素环为母核,通过引入磺酰脲结构,根据孪药的原理合成了一系列降血糖的香豆素衍生物[1],其中代号为SU-118的新化合物(见结构图)经药理实验显示具有良好的降血糖作用。由于SU-118是由两个有效基团拼合而成,而这两个有效基团的降糖机制并不相同,所以这个新化合物完全有可能同时兼备两种机制或者在体内有新的作用机制从而使其降糖作用明显。因此,该化合物有望成为一种不同于磺酰脲类药物的新型降血糖药物。研究SU-118与血清白蛋白的相互作用对理解此类药物的作用机理以及进行药物筛选具有重要意义。
1 实验部分
本文采用分子对接方法进行模拟实验,该方法是指在分子模拟环境中,两个或两个以上的分子模型通过几何形状,化学环境以及能量的匹配形成最佳结合的技术。准确地分子对接是研究分子作用机理和设计分子的基础。为了尽可能保证其可靠性,共采用了两种模拟软件,分别是ICM和CAChe软件。
ICM软件的主要功能是将柔性的配体分子对接在能量最佳的位置上。这个能量包含着基于ECEPP/3力场下的配体的内部能量,同时还有范得华力,氢键,静电和疏水的配体或受体的相互作用。ICM程序在内在的调整时间内是用来发现能量最小的位置。每一个运算规则的步骤都是由遵循局部能量最小化原则的两种类型(扭转和位置)的任意构像改变组成的。扭转移动包括完全随机的转变一个任意的扭转角。位置移动包括整个配体作为整体进行类似于布朗运动的平移和旋转。ICM运用梯度最小化分析,发现能量的局部最小化比单一最小化和随机的独自搜索进行的更快。为了改进集中,可以多次从不同位置开始运行。ICM的VLS得分功能是由配体内部的力场能量和配体与受体相互作用的能量组成的。而后者包括范德华力,基于已接近溶剂的表面的疏水作用,静电溶剂化作用和氢键作用。
CAChe软件的主要功能是运用遗传算法(GA)来使小分子对接入蛋白质分子。这种遗传算法是基于已知蛋白质库里的蛋白质与配体的结构信息来推测两个分子的可能嵌入方式。
2 结果与讨论
已有的光谱分析实验结果表明SU-118应键合在HSA的ⅡA和ⅢA位置,通过Cache软件观察ⅡA和ⅢA位置的蛋白质表面确有可供药物分子进入的缝隙,在人血清白蛋白上仅有几个专一的药物键合点,最主要的是位点Ⅰ和位点Ⅱ[2]。位点Ⅰ是华法令和保泰松等类似物的键合位点,也叫华法令键合位点,它定位于HSA上靠近色氨酸214的亚结构域ⅡA区的疏水内腔中,而安定和arylpropionic acid等主要键合于位点Ⅱ,位于HSA上的亚结构域ⅢA区的疏水内腔中,位点Ⅱ也叫苯并二氮/吲哚键合位点。
若是在ⅡA位置上进行分子对接,定义的活性位置如下图所示:
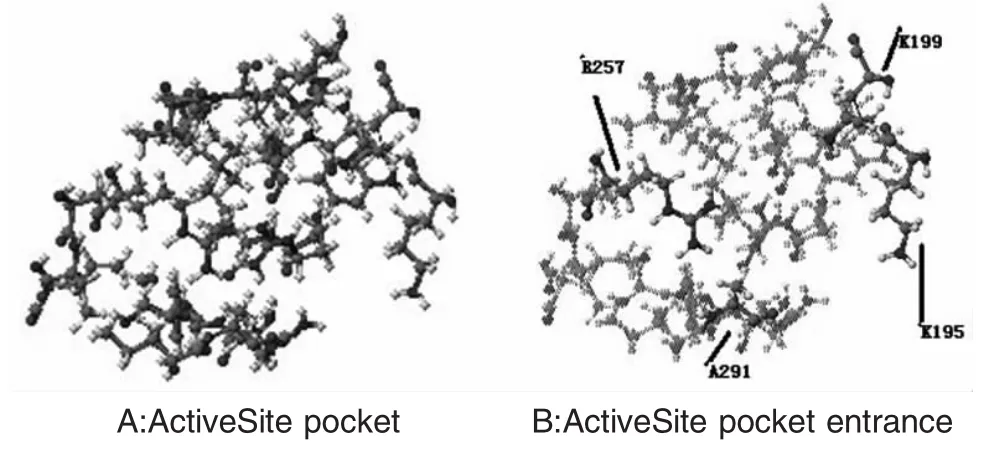
其中含有21个残基K195,K199,F211,W214,A215,V216,R218,L219,R222,F223,V235,L238,V241,H242,R257,L260,A261,I264,S287,I290,A291。这些残基中有四个基团(R218,R222,H242,R257)是亲水基团,底部属于ⅡA位置上的疏水腔。其入口处有残基K195,K199,R257,A291。
用药物分子SU-118进行分子对接实验,结果如图:

Docking Score为-106.998kcal/mole。小分子对接后,其苯环位置先进入腔内,位于疏水的口袋底部,香豆素环部分则位于口袋口附近。SU-118与1N5U上的残基W214的最近距离为3.504A。
HSA分子上有三个内在芳环荧光团:色氨酸﹑酪氨酸和苯丙氨酸,其中色氨酸的荧光变化常用来探测HSA构象变化的信息[3]。色氨酸荧光的猝灭,表明在SU-118-HSA体系中,SU-118在色氨酸附近键合,色氨酸被引入更亲水的环境。在HSA分子上只有一个定位于亚结构域ⅡA疏水口袋中的214位色氨酸,其荧光的猝灭可能是由于SU-118键合于亚结构域ⅡA的疏水口袋附近而引起的。而模拟实验结果得到SU-118与W214的距离为3.504A,而探针与W214的距离为4.970A,可以看出SU-118比探针更加接近W214。由得分看出,丹酰胺分子对接后得分仅有-69.863kcal/mole,而SU-118的得分为-106.998kcal/mole,docking的效果比丹酰胺好很多,由此可以推断出SU-118应该比较容易取代探针键合在ⅡA位置,嵌入ⅡA位置的疏水腔中。
如果在ⅢA位置进行分子对接实验,定义的活性位置如下:
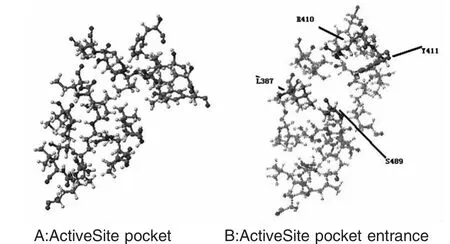
一共有25个残基S342,V344,L345,R348,P384,L387,I388,N391,C392,F403,L407,R410,Y411,L430,G431,V433,G434,C438,M446,A449,E450,L453,R485,P486,S489。其中有 R348,N391,R410,E450,R485五个残基是亲水基团。入口处周围有四个残基R410,Y411,L387,S489。用SU-118进行分子对接实验得分为-78.096kcal/mole。
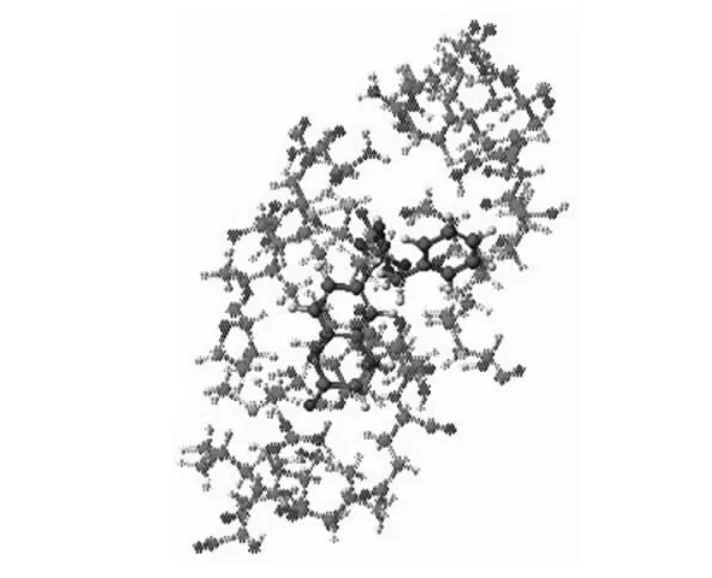
在ⅢA位置进行docking与在ⅡA位置类似,SU-118与丹酰肌氨酸也为竞争性键合,SU-118的得分为-78.096kcal/mole,而丹酰肌氨酸的只有-47.328kcal/mole,由此可见SU-118可以取代丹酰肌氨酸的位点,即HSA上的位点Ⅱ。同样是小分子嵌入疏水腔中,由得分可知,小分子结合在ⅡA位置得分为-106.998kcal/mole,而结合在ⅢA位置得分为-78.096kcal/mole,所以在ⅡA位置小分子结合得比在ⅢA位置更好,由此可知SU-118对位点Ⅰ的亲和力更强。
3 结论
通过模拟实验可以知道,药物分子SU-118在ⅡA位置与HSA键合可以取代探针丹酰胺,在ⅢA位置键合可以取代丹酰肌氨酸,并且SU-118对ⅡA位置的亲和力比对ⅢA位置更强。
[1]Han,Y.,Tu,S.Z.,Zhou,W.et al.J.Chin.Pharm.Univ[J].2002,33,93.
[2]Sudlow,G.,Birkett,D.J.,Wade,D.N.Mol.Pharmacol[J].1975,11,824.
[3]Trynda-Lemiesz,L.,Keppler,B.K.,Koztowski,H.et al.J.Inorg.Biochem[J].1999,73,123.
猜你喜欢
北方牧业(2022年9期)2022-11-22
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
动物营养学报(2018年12期)2018-12-13
材料科学与工程学报(2016年4期)2017-01-15
合成化学(2015年4期)2016-01-17
池州学院学报(2015年3期)2016-01-05
天津科技大学学报(2015年2期)2015-08-09
饲料博览(2014年11期)2014-05-04
无机化学学报(2014年6期)2014-02-28
