
宝儿康颗粒的质量标准研究
2014-04-24 05:18胡军林顾承刚吴
中国医药指南 2014年16期
胡军林顾承刚吴 宁
(1 湖北省食品药品检验监督研究院,湖北 武汉 430064;2 武汉药谷科技开发有限公司,湖北 武汉 430070)
宝儿康颗粒的质量标准研究
胡军林1顾承刚2吴 宁2
(1 湖北省食品药品检验监督研究院,湖北 武汉 430064;2 武汉药谷科技开发有限公司,湖北 武汉 430070)
目的建立宝儿康颗粒的质量控制方法。方法用薄层色谱法对宝儿康颗粒中的太子参、甘草、白术、薏苡仁、山楂进行定性鉴别;以橙皮苷为对照品,采用HPLC法测定了宝儿康颗粒中陈皮的含量。色谱柱:Inertsil ODS-3(不锈钢柱4.6 mm×250 mm,5 μm);流动相:甲醇-0.5%冰醋酸溶液(40∶60);检测波长:283 nm。结果太子参、甘草、白术、薏苡仁、山楂均能以薄层色谱鉴别;含量测定中橙皮苷线性范围0.054~0.54 μg(r=0.9999),回收率为99.2%(RSD=0.63%,n=6)。结论方法操作简便,灵敏度高,专属性和重线性好,可有效地控制宝儿康颗粒的质量。
宝儿康颗粒;橙皮苷;反相高效液相色谱法;太子参;甘草;白术;薏苡仁;山楂
宝儿康颗粒由太子参、茯苓、薏苡仁、白术等十四味中药组成,由宝儿康糖浆改变制剂成型工艺而成,具有补气健脾,开胃消食,渗湿,止泻功效。用于小儿脾胃虚弱,消化不良,食欲不振,大便异常,精神困倦,睡眠不安,夜惊、夜啼等症。原糖浆剂收载于中药部颁标准第十册,标准编号WS3-B-1967-95[1]。宝儿康糖浆保持原剂型的提取工艺不变,仅改变制剂成型工艺,将其制成宝儿康颗粒。为使新制剂质量可控,本文研究制定了本品质量标准。
1 仪器及试剂
高效液相色谱仪(北京创新通恒科技有限公司),型号P3000,检测器3000UV;CQ250超声波仪(上海超声波仪器厂),硅胶G(青岛海洋化工厂),羧甲基纤维素钠(国药集团化学试剂有限公司)。宝儿康颗粒由武汉药谷科技开发有限公司提供(批号20080601、20080602、20080603、20080604、20080701、20080702);橙皮苷对照品(批号110721-20050,供含量测定用)、太子参对照药材(批号1004-200203,供鉴别用)、甘草对照药材(批号121303-200301,供鉴别用)、白术对照药材(批号120925-200407,供鉴别用)、薏苡仁对照药材(批号121254-200307,供鉴别用)、山楂对照药材(批号121138-200403,供鉴别用)均由中国食品药品检定研究院提供,甲醇为色谱纯,水为重蒸水,其他试剂均为分析纯。
2 方法与结果
2.1 太子参薄层色谱鉴别[2]
取本品20 g,研细,加甲醇50 mL,加热回流30 min,滤过,滤液蒸干,残渣加水20 mL使溶解,通过D101型大孔吸附树脂柱(内径1.2 cm,柱高15 cm),用水200 mL、20%乙醇100 mL依次洗脱,弃去水及20%乙醇洗脱液,再用40%乙醇及乙醇各100 mL洗脱,分别收集各自洗脱液,40%乙醇洗脱液预留备用,乙醇洗脱液置水浴上蒸干,残渣加甲醇1 mL使溶解,作为供试品溶液。另取不含太子参的自制样品20 g,同供试品溶液制法制成阴性对照溶液。取太子参对照药材3 g,加水煎煮2 h,离心,取上清液,通过D101型大孔吸附树脂柱,用水200 mL、20%乙醇100 mL洗脱,弃去水及20%乙醇洗脱液,再用乙醇100 mL洗脱,收集洗脱液,蒸干,残渣加甲醇1 mL使溶解,制成对照药材溶液。照《中华人民共和国药典》2010年版一部附录VIB薄层色谱法试验,吸取上述三种溶液各10 µL,分别点于同一硅胶G薄层板上,以甲苯-醋酸乙酯(4∶1)为展开剂,展开,取出,晾干,喷洒1%香草醛硫酸溶液,在105 ℃烘烤至斑点显色清晰。结果在与对照药材色谱相应的位置上,供试品溶液显相同颜色的斑点,阴性样品溶液无斑点显现,对结果无干扰。见图1。

图1 太子参TLC(1 阴性对照;2 太子参对照药材;3、4、5样品)
2.2 甘草薄层色谱鉴别[2]
取2.1项下预留的40%乙醇洗脱液,置水浴上蒸干,加甲醇1 mL溶解残渣,作为供试品溶液。另取不含甘草的自制样品20 g,同供试品溶液的制法配制成阴性对照溶液。再取甘草对照药材0.5 g,加乙醚40 mL,加热回流1 h,滤过,弃去乙醚液,药渣加甲醇30 mL,加热回流1 h,滤过,滤液蒸干,加水30 mL溶解残渣,用正丁醇萃取3次,每次20 mL,合并正丁醇液,用水洗涤3次,正丁醇液蒸干,加甲醇1 mL溶解残渣,制成对照药材溶液。照《中国药典》2010年版一部附录VI B薄层色谱法试验,吸取上述供试品溶液、阴性对照溶液各10 µL和对照药材溶液5 µL,分别点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,以醋酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)为展开剂,展开,取出,晾干,喷洒10%硫酸乙醇溶液,在105 ℃烘烤至斑点显色清晰。结果在与对照药材色谱相应的位置上,供试品溶液显相同颜色的斑点,阴性样品溶液无斑点显现,对结果无干扰。见图2。

图2 甘草TLC(1.阴性对照;2.甘草对照药材;3、4、5.样品)
2.3 白术薄层色谱鉴别[2]
取本品25 g,研细,加甲醇50 mL加热回流30 min,滤过,滤液蒸干,残渣加水25 mL使溶解,用石油醚(60~90 ℃)振摇萃取3次,每次20 mL,合并石油醚液,水液预留备用,石油醚液挥干,加石油醚(60~90 ℃)1 mL溶解残渣,作为供试品溶液。另取不含白术的自制样品25 g,同供试品溶液的制法制成阴性对照溶液。再取白术对照药材1.5 g,加水100 mL,煎煮2 h,过滤,滤液浓缩至20 mL,用石油醚(60~90 ℃)振摇萃取3次,同法制成对照药材溶液。照《中华人民共和国药典》2010年版一部附录VI B薄层色谱法试验,吸取上述三种溶液各10 µL,分别点于同一硅胶G薄层板上,以环己烷-醋酸乙酯(7∶3)为展开剂,展开,取出,晾干,喷洒5%对二氨基苯甲醛硫酸溶液,在105 ℃烘烤10 min,置紫外光灯(365 nm)下检视。结果在与对照药材色谱相应的位置上,供试品溶液显相同颜色的斑点,阴性样品溶液无斑点显现,对结果无干扰。见图3。
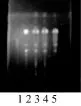
图3 白术TLC(1.阴性对照;2.白术对照药材;3、4、5.样品)
2.4 薏苡仁的薄层色谱鉴别[2]
取不含薏苡仁的自制样品25 g,照2.3项下的供试品溶液制法配制成阴性对照溶液。另取薏苡仁对照药材2 g,加石油醚(60~90 ℃)20 mL,超声处理30 min,滤过,滤液挥至约1 mL,制成对照药材溶液。照《中华人民共和国药典》2010年版一部附录VI B薄层色谱法试验,吸取2.3项下供试品溶液及上述两种溶液各10 µL,分别点于同一硅胶G薄层板上,以石油醚(60~90 ℃)-醋酸乙酯-甲酸(10∶2∶0.1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果在与对照药材色谱相应的位置上,供试品溶液显相同颜色的斑点,阴性样品溶液无斑点显现,对结果无干扰。见图4。
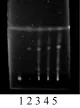
图4 薏苡仁TLC(1.阴性对照;2.薏苡仁对照药材;3、4、5样品)
2.5 山楂薄层色谱鉴别[2]
取2.3项下的水液,用醋酸乙酯萃取2次,每次20 mL,合并醋酸乙酯液,蒸干,加甲醇1 mL溶解残渣,作为供试品溶液。另取不含山楂的自制样品25 g,照供试品溶液制法配制成阴性对照溶液。再取山楂对照药材2 g,加水100 mL,煎煮2 h,滤过,滤液浓缩至20 mL,以稀盐酸调节pH值至1~2,用醋酸乙酯振摇提取2次,同法制成对照药材溶液。照《中华人民共和国药典》2010年版一部附录VI B薄层色谱法试验,吸取上述两种溶液各10 µL,分别点于同一硅胶G薄层板上,以环己烷-醋酸乙酯-甲酸(20∶20∶1)为展开剂,展开,取出,晾干,喷洒2%三氯化铁乙醇溶液,在105 ℃烘烤至斑点显色清晰。结果在与对照药材色谱相应的位置上,供试品溶液显相同颜色的斑点,阴性样品溶液无斑点显现,对结果无干扰。见图5。

图5 山楂TLC(1.阴性对照;2.山楂对照药材;3、4、5样品)
2.6 橙皮苷的含量测定[2]
2.6.1 色谱条件与系统适应性试验:色谱柱:Inertsil ODS-3(不锈钢柱4.6mm×250mm,5 μm);流动相:甲醇-0.5%冰醋酸溶液(40∶60);检测波长:283 nm;柱温:30 ℃;流速:1 mL/min;进样量:10 μL;检测灵敏度:1.0000AUFS;理论塔板数按橙皮苷峰计算应不低于2000。色谱图见图6。
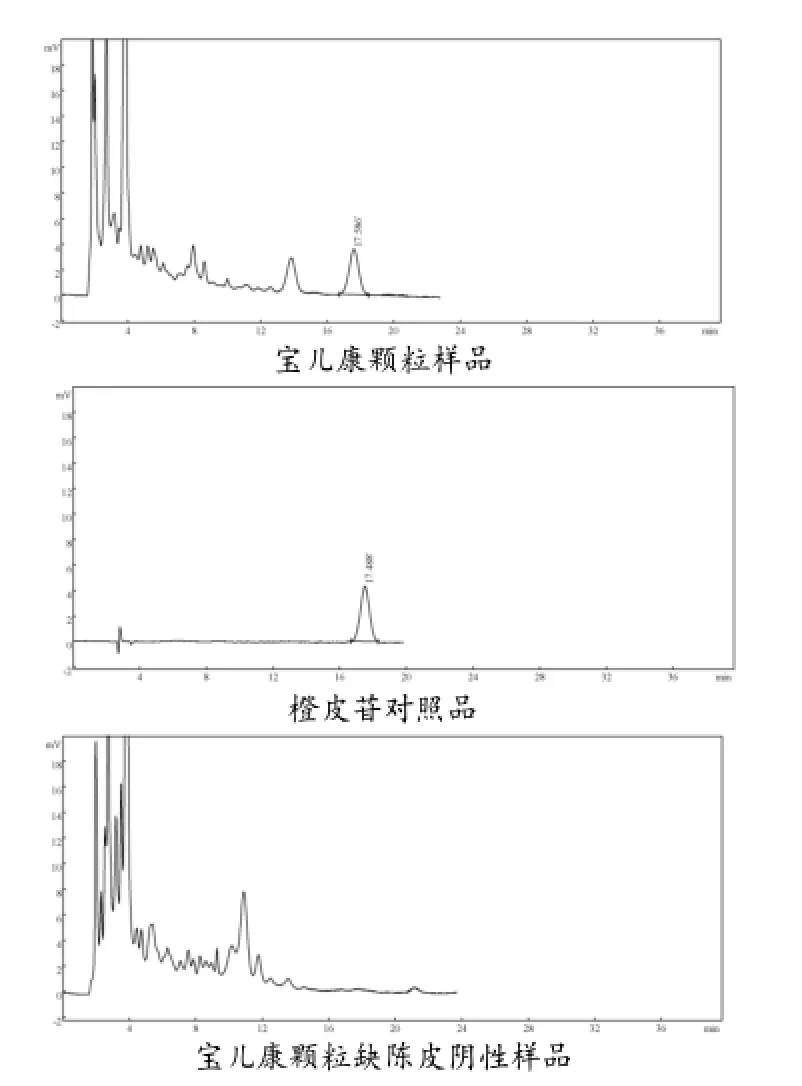
图6 样品、对照品、阴性样品色谱峰
2.6.2 溶液制备:精密称取橙皮苷对照品适量,加甲醇制成25 μg/1 mL的溶液,作为对照品溶液。取本品适量,研细,取约0.7 g,精密称定,置具塞锥形瓶中,精密加入甲醇25 mL,称定重量,置水浴上加热回流1 h,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。另取不含陈皮的自制样品适量,同法制备成阴性对照溶液。
2.6.3 方法学考察
线性关系考察:精密称取橙皮苷对照品5.40 mg,置100 mL量瓶中,用甲醇溶解并稀释至刻度,制成0.0540 mg/mL的溶液。分别精密吸取上述对照品溶液1、2、3、5,7,10μL,分别进样,测定。以横坐标为橙皮苷的量,以纵坐标为峰面积,测得回归方程y=723804.4x-2437.04,r=0.9999。结果表明橙皮苷进样量在0.054~0.54μg范围内与峰面积线性关系良好。
精密度试验: 精密吸取同—份对照品溶液10 μL,按色谱条件,重复进样6次,测定峰面积,结果RSD=0.62%,表明仪器精密度良好。
供试品溶液稳定性试验:分别精密吸取同—份供试品溶液10 μL,分别在0、2、4、6、8、10 h进样,测定峰面积,结果RSD为0.69%(n=6)。结果表明供试品溶液在10 h内保持稳定,能满足测定的时间要求。
重复性试验:取同一批(批号为20080601)样品6份,按供试品溶液制备方法提取制备并测定,计算含量,测得平均值为0.79 mg/g,RSD(%)=1.06%,表明方法的重现性良好。
准确度试验:取已知含量的样品(批号为20080601,含量为0.79 mg/g)6份,分别精密加入浓度为12.1 μg/μL的橙皮苷对照品溶液各25 μL(即0.3025mg),照供试品溶液制法制备溶液,测定含量,计算回收率。结果见表1。
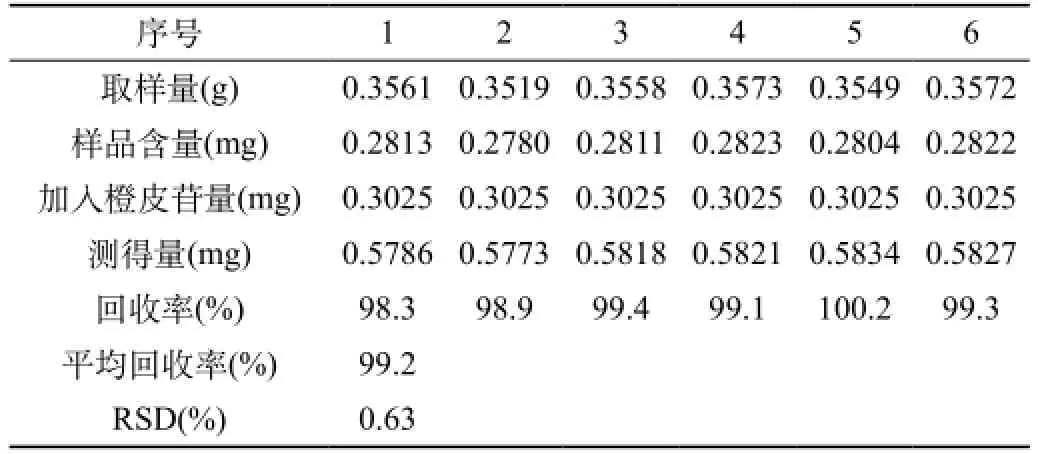
表1 加样回收率试验结果
表1结果表明,本方法的回收率良好。
2.6.4 样品含量测定
取供试品溶液和对照品溶液各10μL,注入高效液相色谱仪,记录峰面积,按外标法计算橙皮苷含量,结果见表2。

表2 样品含量测定结果
3 讨 论
在太子参、甘草的薄层鉴别中将样品通过D101型大孔吸附树脂柱(内径1.2 cm,柱高15 cm),制得样品溶液颜色较浅,杂质较少,斑点清晰,分离效果好。鉴别项阴性对照均无干扰,且操作简便,重现性好。
在含量测定项下,参考《中华人民共和国药典》2005年版一部健胃消食片含测项下橙皮苷含测色谱条件及样品制备方法,峰形良好,与其他色谱峰分离度符合要求。阴性对照对本品含量测定无干扰。通过研究,本文为宝儿康颗粒的生产提供了可行的技术方法。
[1] 国家药典委员会.中华人民共和国卫生部药品标准中药成方制剂第十册[S].1995:116.
[2] 国家药典委员会.中国药典.一部[S].北京:化学工业出版社,2005:59-567.
R282.7
B
1671-8194(2014)16-0094-03
猜你喜欢
现代农业科技(2022年3期)2022-02-21
花卉(2021年14期)2021-07-26
农技服务(2020年6期)2020-07-01
幸福·健康版(2018年3期)2018-03-23
幸福(2018年9期)2018-01-25
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
海峡科技与产业(2016年3期)2016-05-17
消费导刊(2016年3期)2016-04-13
中国当代医药(2015年9期)2015-03-01
中国当代医药(2015年8期)2015-03-01
