
基于单池药物溶出/吸收仿生系统评价格列吡嗪不同制剂的释药特征及其体内外相关性*
2014-04-20 08:17:12寻明金YaroPeter谷升盼
天津中医药 2014年2期
寻明金,Yaro Peter,谷升盼,何 新,2
(1.天津中医药大学中药学院,天津 300193;2.天津市现代中药重点实验室,天津 300193)
·中药研究·
基于单池药物溶出/吸收仿生系统评价格列吡嗪不同制剂的释药特征及其体内外相关性*
寻明金1,Yaro Peter1,谷升盼1,何 新1,2
(1.天津中医药大学中药学院,天津 300193;2.天津市现代中药重点实验室,天津 300193)
摘要:[目的]考察格列吡嗪片及其缓释片在单池药物溶出/吸收仿生系统(Single cell-DDASS)的释放特征,以及与比格犬体内吸收程度的相关性。[方法]采用《中华人民共和国药典》(以下简称《药典》)溶出度法和Single cell-DDASS法对格列吡嗪片及其缓释片进行体外释放度实验,用Wagner-Nelson方程计算不同格列吡嗪制剂在体内的吸收百分数,并与相应时间点体外累积释放度进行线性回归,进一步考察其体内外相关性(IVIVC)。[结果]格列吡嗪缓释片体外释放度的最优拟合方程是一级动力学方程,释放机制为溶蚀机制。格列吡嗪片在《药典》溶出法中释放过快,无法建立体内外相关性,而Single cell-DDASS法中累积释放度与比格犬体内吸收百分数之间存在相关性(r=0.773 2,P<0.05)。格列吡嗪缓释片在《药典》溶出法中累积释放度与比格犬体内吸收百分数之间密切相关(r=0.811 5,P<0.05)。格列吡嗪缓释片在Single cell-DDASS法中累积释放度与比格犬体内吸收百分数之间极密切相关(r=0.987 7,P<0.01)。[结论]格列吡嗪片及其缓释片在Single cell-DDASS法中累积释放度与比格犬体内吸收百分数之间的相关性明显优于《药典》溶出法释放百分数与比格犬体内吸收百分数的相关性,表明Single cell-DDASS能够良好预测格列吡嗪不同制剂在体内的动力学特征。
关键词:单池药物溶出/吸收仿生系统;格列吡嗪;体内外相关性
生物药剂学分类(BCS)中的BCS II类药物(低溶解性、高渗透性),其药物溶解度低,并且溶解度易受pH的影响[1],药物释放是体内药物吸收的限速步骤。2010年版《中华人民共和国药典》(以下简称《药典》)规定的药物溶出度测定装置包括转篮法、浆法、小杯法[2],主要是通过搅拌或旋转在单一溶出介质中产生强制对流对溶出的药物进行混合,其局限性在于无法考察药物制剂从胃(pH 1~2)移行至肠(pH 5~8)过程中急剧的pH动态变化对药物溶解性、稳定性以及药物释出的影响,得到的是药物制剂的累积溶出度,无法动态的考察其实时溶出特征。药物溶出/吸收仿生系统(DDASS)是近年发展起来的连续动态评价药物溶出和跨膜透过特征的新型评价技术。He等[3-4]用数学模型和动力学参数表征药物变化规律的动力学特点,并且研究不同胃酸情况下对药物溶出率和跨膜吸收率的影响。随后,Sugawara等[5]采用该系统成功评价了缓释颗粒的吸收特征。在此基础上,本课题组对该体系进行了改进,用于水溶性药物缓释制剂的释药规律及体内外相关性研究[6-7],以及中药复杂成分配伍规律研究[8]。为了准确评价肠溶性药物制剂的释药规律和吸收特征,本课题组建立了单池药物溶出/吸收仿生系统(Single cell-DDASS)[9],该系统考虑了人体胃肠道液体的缓冲能力、渗透压、表面张力以及胃液肠液pH值的不同,并通过一定时间切换不同pH值药物释放介质可以连续、动态地模拟药物制剂从胃移行至肠道崩解、释放的过程,更真实模拟了体内药物溶出过程。目前该模型的应用研究已受到国内外同行广泛关注。
格列吡嗪是第2代磺酰脲类口服降糖药,主要用于治疗非胰岛素依赖型糖尿病[10]。该药吸收较快且无首过效应。格列吡嗪在水中溶解度为7.79 mg/L,并且溶解度随着pH增大而增大[11],属于典型的BCSⅡ类药物。本研究采用Single cell-DDASS考察格列吡嗪片及其缓释片的释放特征,以及与比格犬体内吸收程度的相关性,为评价BCSⅡ类药物制剂提供一种新技术。
1 材料与方法
1.1 药品和材料 格列吡嗪对照品(批号:100281-200602,纯度:99.4%)、佛手苷内酯标准品(批号:100250-200503,纯度:99%)均购自中国药品生物制品检定所,格列吡嗪片(秦苏 ,扬子江药业集团有限公司,每片5 mg,批号:H10970356),格列吡嗪缓释片(美吡达®,海南赞邦制药有限公司,每片5 mg,批号:H10930076)。水为纯水,甲醇、乙腈为色谱纯,其他试剂均为分析纯。
1.2 实验动物 雄性比格犬6只,清洁级,体质量(8.5±0.5)kg,购于军事医学科学院实验动物中心,许可证编号:SCXK(京)2009-0012。犬膨化饲料购买于北京科奥协立动物饲料有限公司。犬在(25±2)℃,相对湿度45%~65%,光照/黑暗为12 h/12 h条件下饲养,自由饮食、饮水,适应性饲养1周后开始实验。
1.3 仪器
1.3.1 主要仪器 Merrler Toledo AX205十万分之一分析天平(瑞士Mettler Toledo公司),SOTAX AT7 smart溶出仪(瑞士SOTAX公司),Waters 600E自动型高效液相仪(美国Waters公司),PHSJ-4A型实验室pH计(上海精密科学仪器有限公司),Tomos Model 2-16A高速离心机(美国Tomos公司)。
1.3.2 Single cell-DDASS Single cell-DDASS如图1所示,其核心部位由药物溶解室和扩散池两部分组成,药物溶解室置一磁力搅拌器和载药篮,出口部分安装有不锈钢筛网。扩散池部分,在供给室和接收室之间嵌合大鼠离体肠管,使得肠腔浆膜侧为供给室,肠腔黏膜侧为接收室,供给室和接收室中的介质分别是药物溶解液(pH=6.8)和接受液(pH=7.4)。
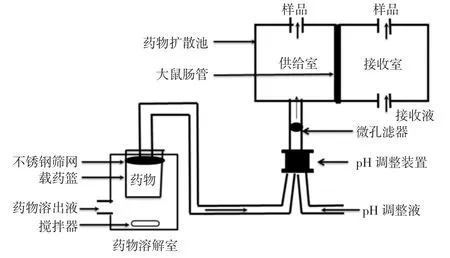
图1 单池药物溶出/吸收仿生系统示意图Fig.1 Schematic diagram of Single cell-DDASS
1.4 分析方法的建立
1.4.1 格列吡嗪体外分析方法的建立
1.4.1.1 色谱条件 TC-C18色谱柱(5 μm,150 mm× 4.6 mm,Agilent,美国),流动相为甲醇-0.1%冰乙酸溶液(57∶43),体积流量1.0 mL/min,检测波长226 nm,柱温30℃,进样量10 μL。
1.4.1.2 专属性 在释放介质中按照格列吡嗪片处方比例加入空白辅料做为空白样品,在空白样品中加入适量格列吡嗪做为对照样品,从溶出系统中随机取样做为待测样品。样品分别经0.45 μm微孔滤膜滤过,按“1.4.1.1”项下条件测定,记录色谱图。
1.4.1.3 线性范围 精密称取格列吡嗪对照品,用流动相制备成900 μg/mL的储备液。将格列吡嗪储备液用流动相稀释制备成30、10、3.33、1.11、0.37、0.12、0.041 μg/mL系列质量浓度,按“1.4.1.1”项下条件测定峰面积,以格列吡嗪质量浓度为横坐标,以峰面积值为纵坐标进行线性回归。
1.4.1.4 准确度和精密度 用溶出介质将格列吡嗪储备液稀释成10.00、1.00、0.10 μg/mL不同浓度的溶液,做为质量控制(QC)样品,经0.45 μm微孔滤膜滤过,按“1.4.1.1”项下色谱条件测定,准确度结果以RE(%)表示。将上述浓度的溶液连续3 d测定,每天连续测定3次,计算各浓度日间、日内精密度RSD(%)。
1.4.1.5 稳定性 吸取同一供试品溶液,分别于0、1、2、4、6、8 h按上述色谱条件进样,每次重复进样5次,测定室温下QC样品的峰面积,通过每个时间点峰面积与0时样品峰面积之比计算剩余百分比。
1.4.2 格列吡嗪体内分析方法的建立
1.4.2.1 色谱条件 TC-C18色谱柱(5 μm,150 mm× 4.6 mm,Agilent,美国),流动相为甲醇-0.1%冰乙酸溶液(50∶50),体积流量1.0 mL/min,检测波长226 nm,柱温30℃,进样量20 μL。
1.4.2.2 专属性 空白血浆做为空白样品,在空白样品中加入一定浓度的格列吡嗪溶液和内标溶液(佛手苷内酯)作为对照样品,从血浆样品中随机取样作为实测样品。同血浆样品处理后,按“1.4.2.1”项下条件测定,记录色谱图。
1.4.2.3 线性范围 比格犬空白血浆500 μL中,分别加入不同量的格列吡嗪和定量的内标液,配置成相当于格列吡嗪为18,9,4.5,2.25,1.13,0.56,0.28 μg/mL的血浆样品,样品经处理后,按“1.4.2.1”项下条件测定峰面积,以待测药物浓度为横坐标,待测药物与内标物峰面积比值为纵坐标进行线性回归。
1.4.2.4 准确度和精密度 比格犬空白血浆500 μL中,依次加入9.00,2.25,0.56 μg/mL的格列吡嗪溶液和定量的内标液,按血浆样品处理后,作为QC样品,每一浓度QC样品平行6次,连续测定3 d,根据当日的标准曲线,计算QC样品的测得浓度。根据QC样品测定结果计算方法的日内、日间精密度和准确度,精密度用RSD(%)表示,准确度用RE(%)表示。
1.4.2.5 提取回收率 同上制备QC样品,每一浓度QC样品平行测定6次。将适量的格列吡嗪与内标加入到空白血浆中,使得理论上与QC样品浓度一致,即得回收率对照溶液,回收率对照溶液与QC样品同法进样,样品色谱图中各色谱峰面积与相应回收率对照溶液对应的峰面积之比,即为绝对回收率。
1.4.2.6 稳定性 将上述9.00、2.25、0.56 μg/mL 3个浓度QC样品进行稳定性考察:经过3次冷冻-冻融循环的稳定性研究。室温放置0、1、2、4、6、8 h的稳定性研究。每个浓度进行3个样本分析。
1.5 体外释放度实验
1.5.1 《药典》溶出法溶出度实验 取格列吡嗪片6片,参照溶出度测定法(《药典》二部附录XC),采用溶出度测定法第一法装置,以500 mL磷酸盐缓冲液(pH 7.4)为释放介质,转速100 r/min,依法操作,格列吡嗪片于0.08、0.17、0.25、0.33、0.42、0.50 h时分别取溶液2 mL,0.45 μm微孔滤膜滤过,并即时在溶出杯中补充同温度的释放介质2 mL,分别取续滤液。按“1.4.1.1”项下色谱条件测定。
格列吡嗪缓释片6片,参照缓释制剂释放度测定法第一法(《药典》二部附录XD),采用溶出度测定法第一法装置(附录XC),以500 mL磷酸盐缓冲液(pH 7.4)为释放介质,转速100 r/min,依法操作,格列吡嗪缓释片于0、1、2、3、4、5、6、8、10、12、24 h时分别取溶液2 mL,0.45 μm微孔滤膜滤过,并即时在溶出杯中补充同温度的释放介质2 mL,分别取续滤液。按“1.4.1.1”项下色谱条件测定。
1.5.2 Single cell-DDASS法释放度实验 使用图1装置进行实验。格列吡嗪片和缓释片的投药量是1片/次(规格:5 mg),每种剂型平行实验3次。将药物制剂投入药物溶解室中开始实验,溶解的药物随药物溶解液流至pH调节装置,继而传送到药物扩散池,由于本次实验目的在于考察溶出过程,供给室和接收室之间被封闭,仅从供给室取样。各药物溶解液的流速由恒速蠕动泵控制在0.5 mL/min;使用样品程控自动收集器收集待测样品,每间隔10min收集1次。整个实验过程中保持各缓冲液温度为37℃。待测样品处理后按照“1.4.1.1”项下色谱条件测定。
1.6 释放模型的拟合 计算不同时刻药物释放度和累积释放度,绘制释放度-时间曲线和累积释放度-时间曲线。SPSS 18.5软件拟合固体制剂体外释药方程,考察该制剂在4种不同释放模型下的拟合方程和拟合优度r。不同释放模型的方程分别如下:
零级动力学方程:Q=at+b
一级动力学方程:ln(1-0.01Q)=at+b
Higuchi方程:Q=at1/2+b
Ritger-Peppas方程:lnQ=alnt+b
式中Q为累积释放度(%),t为时间(min),a,b为常数。
1.7 比格犬体内实验 采用单剂量双周期交叉实验方案,将6只雄性比格犬随机分为甲、乙两组,每组3只,实验前12 h开始禁食。甲组比格犬口服格列吡嗪片,乙组口服格列吡嗪缓释片,经1周以上洗脱期,给比格犬交叉服药。
比格犬取血点设计如下所示:格列吡嗪片为给药前0 h和给药后0.33、0.66、1、1.5、2、2.5、3、6、9、12 h于比格犬前肢静脉取血2 mL。格列吡嗪缓释片为给药前0 h和给药后1、1.5、2、2.5、3、3.5、4、5、6、9、12 h于比格犬前肢静脉取血2 mL。将全血置于1%肝素抗凝的干燥带塞玻璃离心管,以转速4 000 r/min,温度4℃的条件下离心10 min,取上清液置于-20℃冰箱保存,待用。
1.8 血浆样品预处理 取血浆样品500 μL,分别加入浓度为4.89μg/mL的内标溶液(佛手苷内酯)20μL,涡旋混匀后,加入萃取液乙醚4 mL,涡旋3 min。在转速4 000 r/min,温度4℃的条件下离心10 min,取上清液3.5 mL,使用氮气吹干仪在室温下吹干,用100 μL甲醇复溶,涡旋3 min,在转速14 000 r/min,温度4℃的条件下离心10 min,取上清液进样。
1.9 体内数据处理 WinNonlin作为药代动力学和药效学建模及非房室模型分析的行业标准,是目前国外应用最广泛的药代动力学软件。本实验的体内血药浓度数据采用Phoenix WinNonlin version 6.1(Pharsight Co.,Ltd,美国)药动学软件处理,通过非房室模型计算体内药代动力学参数。
1.10 体内外相关性分析 体内吸收呈现单室模型的药物,可换算成体内吸收率—时间的体内吸收曲线,药物的口服吸收率Fa可相应地按Wagner-Nelson方程计算[12],如下:
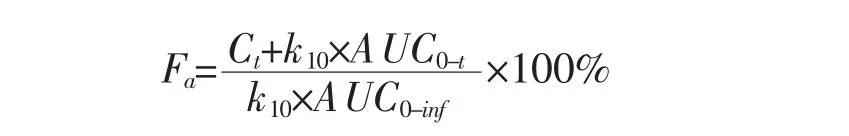
式中:Ct为t时间的血药浓度,k10为消除速率常数,AUC0-t为t时间内药时曲线下面积,AUC0-inf为0到无穷大时间内药时曲线下面积。
利用线性最小二乘法回归原理,将同批试样体外溶出曲线和体内吸收曲线上对应的各个时间点的溶出率和吸收率回归,得直线回归方程及相关系数。其中,X轴为药物累积释放度结果,Y轴为药物体内吸收度结果。
2 实验结果
2.1 格列吡嗪分析方法的验证
2.1.1 格列吡嗪体外分析方法的验证
2.1.1.1 专属性 如图2所示,药物溶解液及辅料对格列吡嗪色谱峰无干扰,专属性良好。
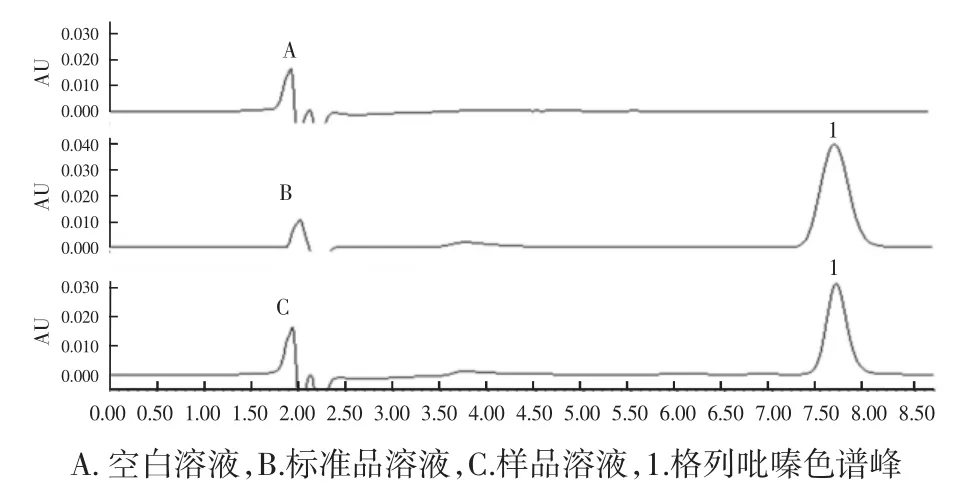
图2 格列吡嗪HPLC色谱图Fig.2 HPLC chromatogram of glipizide
2.1.1.2 线性范围 对照品储备液用流动相逐级稀释,等差制备成一系列浓度,按上述“1.4.1.1”项下进样测定峰面积,以标品浓度(C)为横坐标,以峰面积(A)值为纵坐标进行线性回归,回归方程为Y^=26 751.48X+4.73(r2=0.999 4),表明格列吡嗪在0.041~30 μg/mL范围内线性关系良好。
2.1.1.3 准确度和精密度 格列吡嗪高、中、低浓度的回收率和日内、日间精密度结果如表1所示,表明该方法准确度和精密度皆符合要求。
2.1.1.4 稳定性考察 格列吡嗪高、中、低浓度于不同时间点药物剩余百分比(98.41±0.72)%、(101.77± 1.28)%、(98.83±2.03)%,表明格列吡嗪在溶出介质中8 h内稳定性符合要求。
2.1.2 格列吡嗪体内分析方法的验证
2.1.2.1 专属性 专属性实验结果见图3。由图A、B、C看出:在本实验条件下,空白样品中的内源性杂质对待测药物格列吡嗪和内标物佛手苷内酯的测定无干扰,方法的专一性良好。
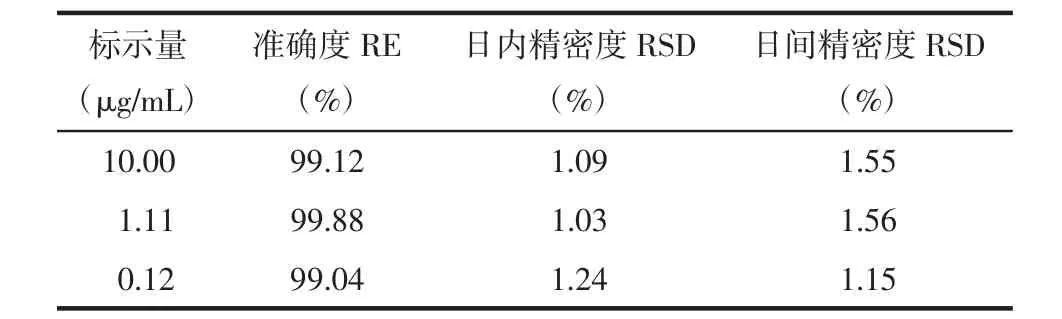
表1 格列吡嗪准确度和精密度测定结果(n=3)Tab.1 Accuracy and precision for the determination of glipizide(n=3)
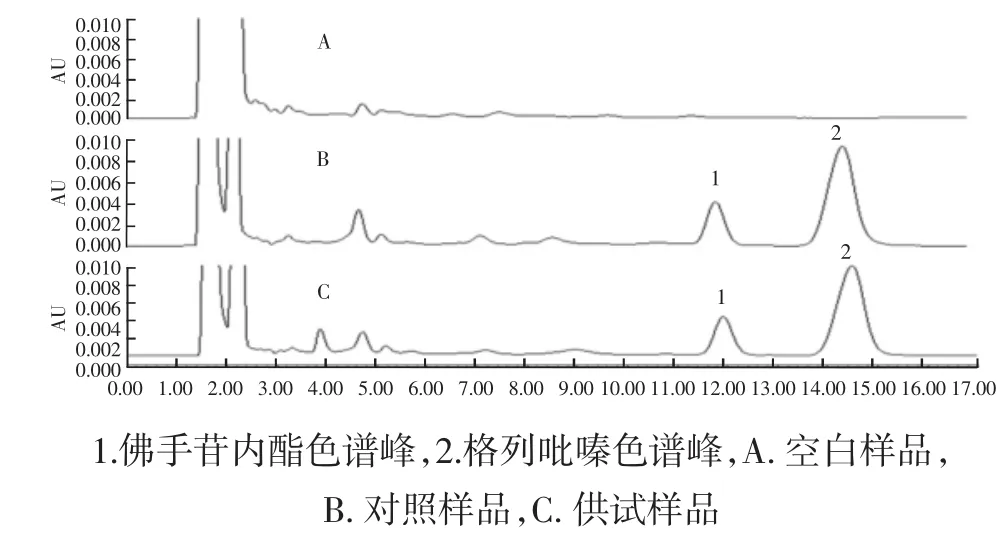
图3 格列吡嗪高效液相色谱图Fig.3 HPLC chromatogram of glipizide and Bergapton
2.1.2.2 线性范围 格列吡嗪系列浓度(X)-峰面积比值(Y)线性回归方程为Y^=0.408 6X-0.074 3(r2=0.999 6),表明药物在0.28~18 μg/mL范围内线性关系良好。
2.1.2.3 准确度和精密度 格列吡嗪的准确度RE和日内、日间精密度RSD结果见表2,表明该方法准确度、精密度均符合要求。
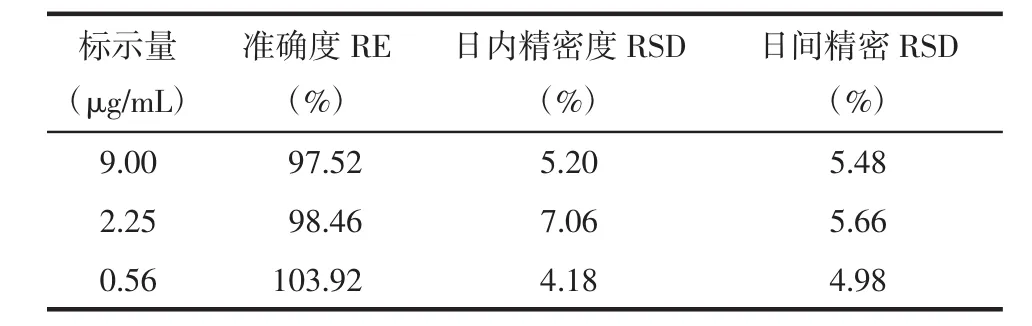
表2 格列吡嗪犬血浆样品的准确度和精密度(n=6)Tab.2 Accuracy and precision of glipizide in plasma of beagle dogs(n=6)
2.1.2.4 提取回收率 格列吡嗪高、中、低浓度的绝对回收率分别为(89.28±5.69)%、(88.99±2.97)%、(83.90±3.47)%。表明该提取方法满足测定要求。
2.1.2.5 稳定性 格列吡嗪高、中、低浓度于-20℃3次冷冻-冻融循环后峰面积的RSD分别为3.22%、4.81%、3.87%,表明药物稳定性符合要求。
2.2 体外释放度实验研究
2.2.1 《药典》溶出法释放度 分别计算格列吡嗪片和格列吡嗪缓释片不同时刻的累积释放度,并绘制累积释放度-时间曲线,如图4所示。按“1.6”项下不同释放模型下的拟合方程和拟合优度r,如表4所示。格列吡嗪片在其《药典》溶出法中30 min时溶出度为(92.83±2.24)%大于80%,符合《药典》对格列吡嗪片溶出度的要求。格列吡嗪缓释片在《药典》溶出法中24 h时释放度为(91.15±0.98)%,符合“《药典》附录XIX D缓释、控释和迟释制剂指导原则”的规定,说明格列吡嗪缓释片符合缓释特征。格列吡嗪缓释片体外释放度最优拟合方程是一级动力学方程,并且格列吡嗪缓释片为圆柱形,Ritger-Peppas方程幂指数n为0.90(n>0.89),表明格列吡嗪缓释片在《药典》溶出法中释放机制为溶蚀机制。
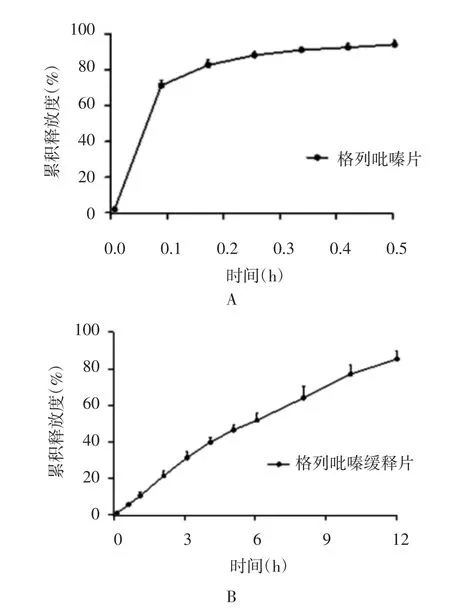
图4 格列吡嗪片(A)和格列吡嗪缓释片(B)在《药典》溶出法中的累积释药曲线(n=6)Fig.4 Cumulative elution profiles of glipizide immediate release tablet(A)and sustained-release tablet(B)in CHP method(n=6)
2.2.2 Single cell-DDASS法释放度 分别计算格列吡嗪片和格列吡嗪缓释片不同时刻的释放度和累积释放度,并绘制相应的释放度-时间曲线和累积释放度-时间曲线,如图5所示。对两种制剂的释放度曲线动力学参数拟合,如表3所示。两种剂型含药物量相同,但是其缓释制剂的Tmax和Cmax与普通片相比有显著性差异(P<0.05),并且缓释片有明显的平台期,表现出明显的缓释作用,说明Single cell-DDASS可以预测不同剂型的释放特征。按“1.6”项下分别进行格列吡嗪缓释片的释放数学模型拟合,在4种不同释放模型下的拟合方程和拟合优度r,如表4所示。从表中可见格列吡嗪缓释片体外释放度最优拟合方程为一级动力学方程,并且格列吡嗪缓释片为圆柱形,Ritger-Peppas方程幂指数n为1.39(n>0.89),表明格列吡嗪缓释片在Single cell-DDASS释放机制为溶蚀机制。

表3 格列吡嗪不同制剂在Single cell-DDASS供给室溶出特征的动力学参数(x±s,n=3)Tab.3 Kinetic parameters of elution into the donor compartment for glipizide dosage forms(x±s,n=3)
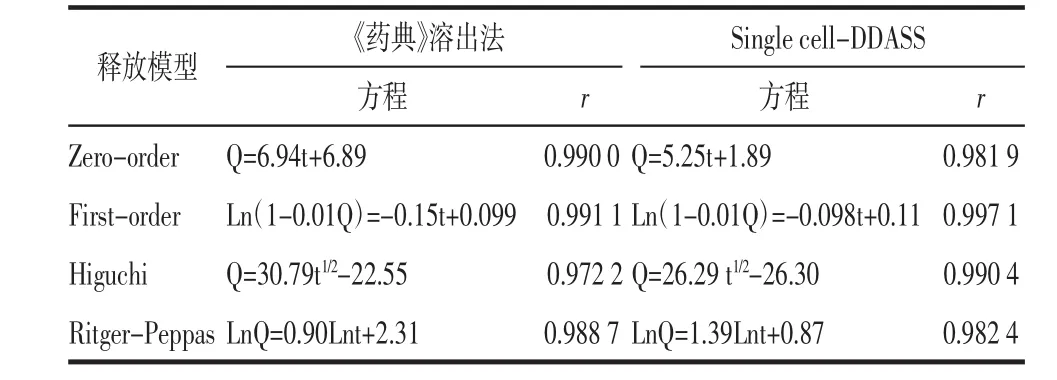
表4 格列吡嗪缓释片在体外释放模型拟合结果Tab.4 Release kinetic model fitting of glipizide sustained release tablet in CHP method and Single cell-DDASS
2.3 比格犬体内药物代谢动力学结果
2.3.1 血浆样品中药物浓度的测定 6只比格犬口服格列吡嗪片和其缓释片后,测定了各时间点血浆样品中格列吡嗪的浓度,平均血药浓度-时间曲线见图6。
2.3.2 药动学参数的计算 格列吡嗪不同制剂在比格犬体内不同时间点血药浓度经WinNonlin药代动力学软件非房室模型拟合之后,求算药代动力学参数如表5所示。
2.4 药物体外释放度与体内吸收程度的相关性研究 格列吡嗪片在《药典》溶出法中30 min溶出度达到80%以上,释放过快,无法建立体内外相关性。格列吡嗪片在Single cell-DDASS法中累积释放度与比格犬体内吸收百分数之间线性回归,其回归方程为:Y^=0.32X-8.41(r=0.773 2,P<0.05)。

图5 格列吡嗪片和格列吡嗪缓释片在Single cell-DDASS中的释放度曲线(A和B)及其相应的累积释药曲线(C和D)Fig.5 Time courses of elution into the donor compartment for glipizide tablet and sustained release tablet in Single cell-DDASS(A and B),and corresponding cumulative elution profiles(C and D)
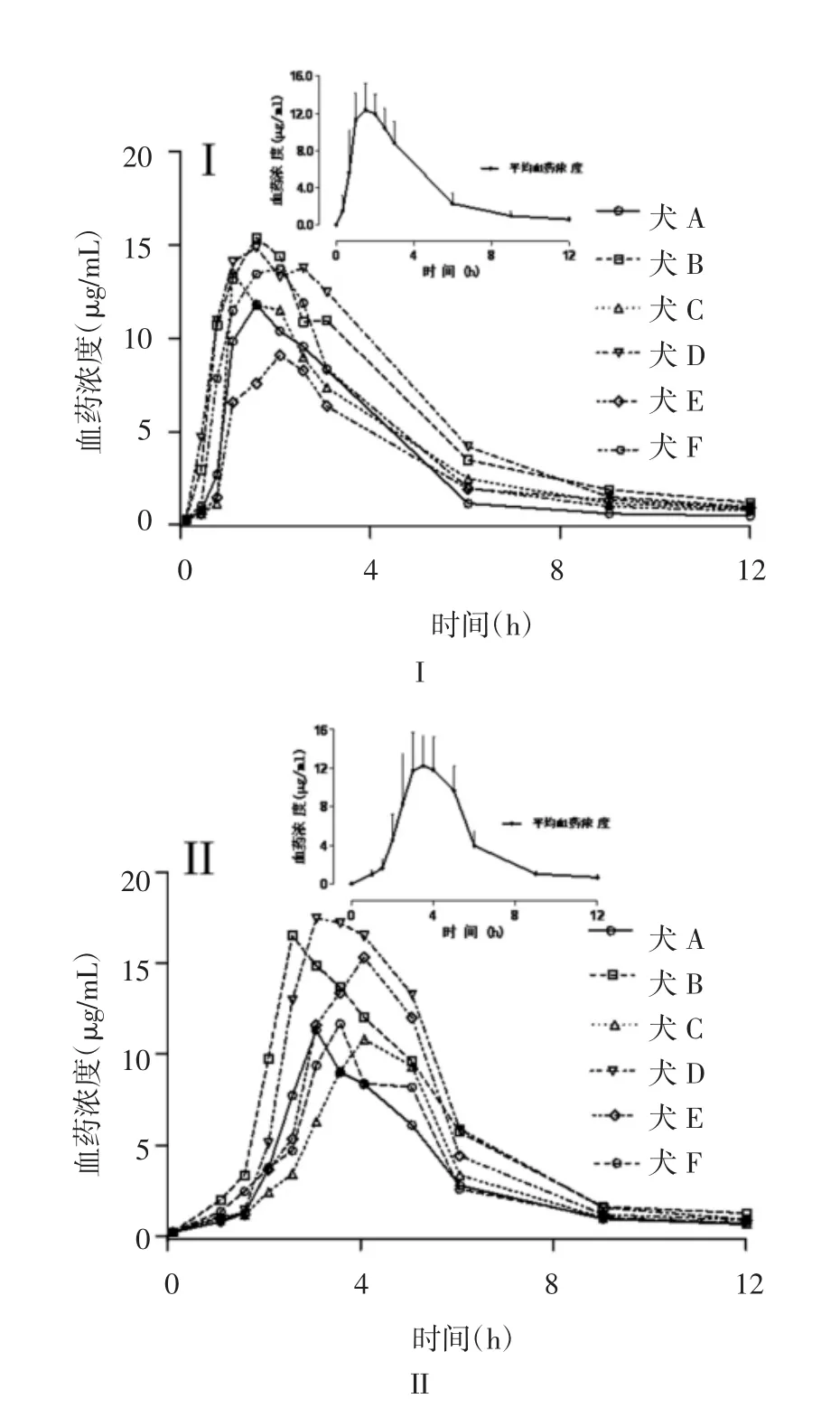
图6 格列吡嗪片(I)及其缓释片(II)在单个比格犬体内药时曲线,单个图表内的小图表示口服给药后犬体内的平均药时曲线(n=6)Fig.6 Profiles of individual plasma concentration after oral administration of glipizide tablet(I)and glipizide sustainedrelease tablet(II)to each beagle dog,and the inserted plot showed the profile of mean plasma concentration after oral administration of drugs(n=6)
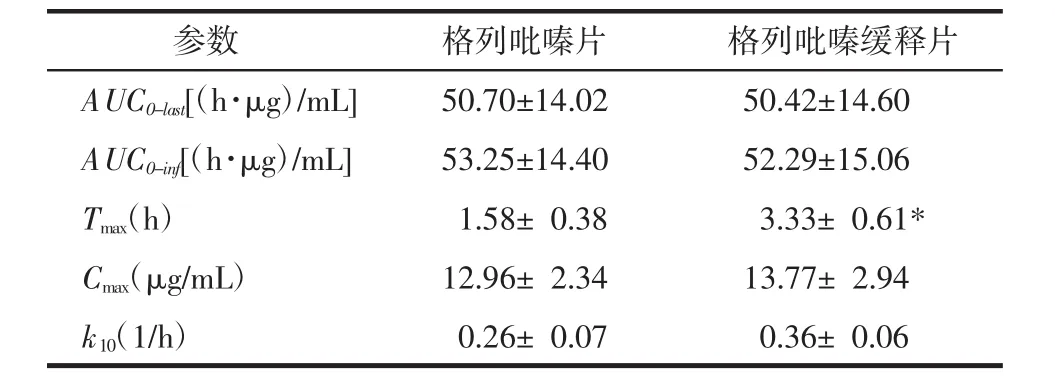
表5 格列吡嗪不同制剂单剂量给予比格犬后体内药代动力学参数(x±s,n=6)Tab.5 Pharmacokinetic parameters of glipizide after a single oral administration of the two tested glipizide dosage forms(x±s,n=6)
格列吡嗪缓释片在《药典》溶出法中累积释放度与比格犬体内吸收百分数之间线性回归,其回归方程为:Y^=0.38X+8.93(r=0.811 5,P<0.05)。而格列吡嗪缓释片在Single cell-DDASS法中累积释放度与比格犬体内吸收百分数之间线性回归,其回归方程为:Y^=0.24X+1.63(r=0.987 7,P<0.01),有极强的相关性。
3 结论
格列吡嗪缓释片体外释放度的最优拟合方程是一级动力学方程,释放机制为溶蚀机制。格列吡嗪片及其缓释片Single cell-DDASS法中累积释放度与比格犬体内吸收百分数之间存在显著的相关性,其相关性明显优于《药典》溶出法累积释放度与比格犬体内吸收百分数的相关性,表明Single cell-DDASS可以用于预测格列吡嗪不同制剂的体内释放及吸收动力学特征。
4 讨论
“《药典》附录XIX D缓释、控释和迟释制剂指导原则”规定,缓释制剂指在规定释放介质中,按要求缓慢地非恒速释放药物,其与相应的普通制剂比较,给药频率比普通制剂减少一半或给药频率比普通制剂有所减少,且能显著增加患者的依从性的制剂。在动物体内药代动力学实验中,与相应的普通制剂相比较,如果缓释制剂Tmax增大,说明该药物制剂释放延缓,则给药时间间隔延长,给药频率有所减少。
表5中所示,格列吡嗪片及其缓释片的Tmax分别为(1.58±0.38)h和(3.33±0.61)h,相比普通制剂,缓释制剂Tmax显著延长(P<0.05),说明格列吡嗪缓释片符合缓释特征。格列吡嗪片及其缓释片的Cmax分别为(12.96±2.34)μg/mL和(13.77±2.94)μg/mL,相比普通制剂,缓释制剂Cmax有所增加,但无显著差异(P>0.05),究其原因可能是犬与人体消化道的结构差异所致。有文献表明:1)药物在人体小肠的转运时间明显长于比格犬。比格犬小肠转运时间约为2~3 h,人体小肠转运时间约为4 h[13-14]。2)比格犬胃肠道的搅拌强度和机械破坏力均强于人体。人体胃肠道内的搅拌强度约相当于体外溶出实验中的浆法10 r/min,比格犬胃肠道内的搅拌强度约相当于体外溶出实验中的浆法100 r/min[15]。禁食状态下人体胃部的机械破坏力约为1.5 N,比格犬胃部的机械破坏力为3.2 N。3)格列吡嗪缓释片的释放机制为溶蚀机制。对于溶蚀机制释放的缓释制剂由于其完整性易被胃肠道破坏,其体内释药速度往往被加快[16-19]。
体内外相关性是用数学模型描述药物体外性质(药物溶出的速率或者程度)与体内特征(血药浓度或药物吸收量)的关系。研究药物制剂的体内外相关性的目的是通过体外溶出度实验,预测药物的体内生物利用度,可指导和优化处方的设计,确立更具代表性的溶出实验规则,合理调整制剂和工艺。通常选择溶出度实验的溶剂尽量接近生理条件[20],才能更好的预测体内吸收情况。Single cell-DDASS的释放介质考虑了人体胃肠道液体的缓冲能力、渗透压、表面张力以及胃液肠液pH值的不同,并通过一定时间切换不同pH值药物释放介质可以连续、动态地模拟药物制剂从胃移行至肠道崩解、释放的过程,真实模拟了体内药物溶出过程[9]。对格列吡嗪两种制剂的在Single cell-DDASS中累积释放百分率和体内累积吸收百分数为考察因素做相关性考察,其相关性明显优于《药典》溶出法累积释放度和体内累积吸收百分数的相关性。表明Single cell-DDASS能够良好预测格列吡嗪不同制剂的体内吸收特征,为评价BCSⅡ类药物制剂提供一种新方法。
参考文献:
[1] Nicolaides E,Galia E,Efthymiopoulos C,et al.Forecasting the in vivo performance of four low solubility drugs from their in vitro dissolution data[J].Pharm Res,1999,16(12):1876-1882.
[2]国家药典委员会编.中华人民共和国药典(二部)[S].2010年版,北京:化学工业出版社,2010:附录73.
[3] Galia E,Nicolaides E,Hörter D,et al.Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs[J].Pharm Res,1998,15(5):698-705.
[4] He X,Sugawara M,Kobayashi M,et al.An in vitro system for prediction of oral absorption of relatively water-soluble drugs and ester prodrugs[J].Int J Pharm,2003,263(1-2):35-44.
[5] Sugawara M,Kadomura S,He X,et al.The use of an in vitro dissolution and absorption system to evaluate oral absorption of two weak bases in pH-independent controlled-release formulations[J].Eur J Pharm Sci,2005,26(1):1-8.
[6] 李自强,刘志东,顾 慧,等.药物溶出/吸收仿生系统研究丹酚B缓释制剂的释放规律[J].药物评价学研究,2010,33(5):367-373.
[7] Li ZQ,He X,Gao X,et al.Study on dissolution and absorption of four dosage forms of isosorbide mononitrate:Level A in vitro-in vivo correlation[J].Eur J Pharm Biopharm,2011,79(2):364-371.
[8] 顾 慧,马叶涛,寻明金,等.应用药物溶出/吸收仿生系统研究三七总皂苷与冰片的配伍规律[J].天津中医药,2012,29(3):284-288.
[9] Liu W,He X,Li Z,et al.Development of a Bionic System for the Simultaneous Prediction of the Release/Absorption Characteristicsof Enteric-Coated Formulations[J].Pharm Res,2013,30(2):596-605.
[10]王长虹.格列吡嗪的药物相互作用[J].中国医院药学杂志,1996, 16(3):205.
[11]杨秀丽,孙 进,何仲贵.格列吡嗪油水分配系数和平衡溶解度的测定[J].中国药剂学杂志,2009,7(3):121-126
[12]Wang Y,Nedelman J.Bias in the Wagner-Nelson estimate of the fraction of drug absorbed[J].Pharm Res,2002,19(4):470-476.
[13]Kabanda L,Lefebvre RA,Van Bree HJ,et al.In vitro and in vivo evaluation in dogs and pigs of a hydrophilic matrix containing propylthiouracil[J].Pharm Res,1994,11(11):1663-1668.
[14]Jennifer BD.Comparison of canine and human gastrointestinal physiology[J].Pharm Res,1986,3(3):123-131.
[15]Katori N,Aoyagi N,Terao T.Estimation of agitation intensity in the GI tract in humans and dogs based on in vitro/in vivo correlation[J]. Pharm Res,1995,12(2):237-243.
[16]Levy G,Leonards JR,Procknal JA.Development of in vitro dissolution tests which correlate quantitantively with dissolution rate-limited drug absorption in man[J].J Pharm Sci,1965,54(12):1719-1722.
[17]Aoki S,Uesugi K,Tatsuishi K,et al.Evaluation of the correlation between in vivo and in vitro release of phenylpropanolamine HCl from controlled-release tablets[J].Int J Pharm,1992,85(1-3):65-73.
[18]Drewe J,Keck M,Guitard P,et al.Relevance of pH dependency on invitroreleaseofbromocriptinefromamodified-releaseformulation[J]. J Pharm Sci,1991,80(2):160-163.
[19]Mojaverian P,Radwanski E,Lin CC,et al.Correlation of in vitro release rate and in vivo absorption characteristics of four chlorpheniramine meleate extended-release formulations[J].Pharm Res,1992, 9(4):450-456.
[20]任晓文,徐为人,连潇嫣,等.水飞蓟宾卵磷脂分散体胶囊剂溶出度测定方法的研究[J].天津中医药,2004,21(1):65-67.
(本文编辑:高 杉,于春泉)
中图分类号:R969.1
文献标识码:A
文章编号:1672-1519(2014)02-0102-08
收稿日期:(2013-08-23)
*基金项目:教育部“长江学者和创新团队发展计划”资助项目(.IRT0973),天津市自然科学基金重点项目(12JCZDJC26100),国家重点基础研究发展计划(973计划)资助项目(2012CB724001)。
作者简介:寻明金(1986-),男,硕士研究生,主要研究方向为药物代谢动力学。
通讯作者:何 新,E-mail:hexintn@163.com。
Studies on release characteristics of different preparation of glipizide and intrinsic and exoteric correlation based on Single cell drug dissolution/absorption simulating system
XUN Ming-jin1,Yaro Peter1,GU Sheng-pan1,HE Xin1,2
(1.Faculty of Chinese Materia Medica,Tianjin University of TCM,Tianjin 300193,China;2.Tianjin State Key Laboratory of Modern Chinese Medicine,Tianjin 300193,China)
Abstract:[Objective]To study the drug release characteristics in Single cell drug dissolution/absorption simulating system(Single cell-DDASS)and the correlation between the dissolution in vitro and the absorption in vivo of glipizide immediate release tablet and glipizide sustained release tablet.[Methods]The release characteristics of glipizide different preparation were investigated using dissolution testing described in Pharmacopoeia of the People’s Republic of China(CHP)and Single cell-DDASS.Single-dose pharmacokinetic studies for the tablets were carried out in six beagle dogs.The percent in vivo absorption in beagle dogs was calculated by Wagner-Nelson method.The correlations between release characteristics in both CHP method and Single cell-DDASS and in vivo absorption were investigated.[Results]The release pattern of glipizide sustained release tablet in vitro could be described by zero-order kinetics and release mechanism was attributed to the dissolution mechanism.Glipizide immediate release tablet dissolution was fast in CHP method and in vitro/in vivo correlation could not be established.The correlation,however,between glipizide immediate release tablet release characteristics in Single cell-DDASS and the absorption in beagle dogs was significant(r=0.773 2,P<0.05).The correlation between release characteristics of glipizide sustained release tablet in CHP method and the absorption in beagle dogs was significant(r=0.811 5,P<0.05);the correlation between release characteristics of glipizide sustained release tablet in Single cell-DDASS method and the absorption in beagle dogs was also significant(r=0.987 7,P<0.01).[Conclusion]There is a significant correlation between Single cell-DDASS release and absorption of beagle dogs for glipizide immediate release tablet and glipizide sustained release tablets,and the correlation degree is higher than that between dissolution test described in CHP method and absorption in beagle dogs.Single cell-DDASS can be used to evaluate the in vivo absorption of glipizide immediate release tablet and sustained release tablet.
Key words:Single cell drug dissolution/absorption simulating system;glipizide;in vitro/in vivo correlation
