
浙江省副溶血性弧菌O3∶K6血清型菌株多位点序列分型研究
2014-04-09 00:58:00梅玲玲张俊彦张云怡
中国人兽共患病学报 2014年3期
梅玲玲,龚 璞,占 利,张俊彦,张云怡
副溶血性弧菌为沿海地区引发食物中毒的主要病原菌[1-2],日本每年由副溶血性弧菌引起的食源性病例约为2万例[3]。我国台湾地区由副溶血性弧菌引起的疾病占细菌性食源性疾病的半数以上[4],内陆1992-2001年间的微生物性食源性疾病暴发事件中由副溶血性弧菌引起的占31.1%[5]。2007年浙江省77起食物中毒事件中,由副溶血性弧菌引发的为35起,792例,占发病总起数和总人数的45.45%和55.00%,占微生物性食物中毒的68.6%、69.3%[6]。副溶血性弧菌引发的食源性疾病对社会稳定、人民健康造成了比较大的危害。
多位点测序分型技术(Multi locus sequence typing,MLST)是为研究菌群基因结构而设计的一种高分辨率分型技术[7]。该方法通过对多个管家基因核酸序列的分析,依据等位基因的多样性,确定菌株的序列型,非常适用于流行病学研究,并可通过互联网上的数据库对不同国家、地区分离的菌株进行比较,至今已成功应用于多种病原体的研究。本文针对浙江省不同来源样品中分离的副溶血性弧菌O3∶K6血清型菌株进行多位点序列分型,通过与PubMLST数据库中菌株基因型比对,分析不同来源分离菌株间的亲缘关系,以掌握浙江省不同来源的副溶血性弧菌O3∶K6血清型菌株在遗传进化间的关系,为开展副溶血性弧菌分子分型研究,制定有针对性的预防控制措施提供理论依据。
1 材 料
1.1菌种来源 62株副溶血性弧菌O3∶K6血清型菌株为本中心保存菌株,其中30株分离于不同地区的门诊腹泻病人,21株分离于15起食物中毒事件的病人,7株分离于食品污染物,4株分离于市售食品。11株分离于2005年,2株分离于2007年,35株分离于2008年,12株分离于2009年,2株分离于2010年。所有菌株经VITEK全自动微生物鉴定分析系统鉴定符合副溶血性弧菌的生化特性。用日本生研株式会社生产的抗血清鉴定为O3∶K6血清型。神奈川试验结果均为阳性。
1.2实验仪器、试剂 5331型梯度PCR仪为德国Eppendorf公司产品; BIO-RAD T2A型全自动数码成像仪分析系统由美国 BIO-RAD科技公司生产;高保真TaqDNA聚合酶PCR试剂盒购自宝生物工程有限公司。
2 方 法
2.1MLST目标基因的选择和引物的设计与合成 选择副溶血性弧菌dnaE、gyrB、recA、dtdS、pntA、pyrC及tnaA7个管家基因作为MLST分析基因。PCR扩增和测序引物引用http://pubmlst网站上发表的副溶血性弧菌PCR引物序列(表1),由上海生工生物工程有限公司合成。引物在使用前将其稀释到25 μmol/L,-20 ℃保存备用。

表1 用于MLST分析的7对引物实验参数
*:上游引物中tgtaaaacgacggccagt部分,下游引物中caggaaacagctatgacc部分为通用测序引物序列部分。
Note: Universal sequencing primer sequences fortgtaaaacgacggccagtin forward primers andcaggaaacagctatgaccin the reverses.
2.2DNA提取 挑取单个菌落悬浮于3 mL 无菌ddH2O中,用麦氏浊度仪调至1个麦氏单位,吸取1 mL转移至Eppendorf管,12 000 r/min离心5 min,弃上清,加1 mL 无菌ddH2O,振荡均匀,100 ℃煮沸15 min,12 000 r/min离心10 min,取上清液作为反应模板, 4 ℃保存备用。
2.3MLST分型
2.3.1PCR扩增靶基因片段 50 μL的PCR反应体系包括:ddH2O 33.25 μL, Premix Ex TaqTM(10×) 5 μL,TaKaRa Taq DNA聚合酶(5 U/μL) 0.5 μL、MgCl2(25 mmol/L) 3.25 μL,dNTP 4 μL,上、下游引物 (25 μmol/L)各0.5 μL,模板DNA 2.0 μL;PCR反应条件:96 ℃预变性5 min; 96 ℃变性1 min,59 ℃~57 ℃退火1 min,72 ℃延伸1 min,30次循环后72 ℃延伸10 min。扩增产物在加入EB(溴化乙锭)的1.5 %琼脂糖凝胶上电泳,紫外灯下观察扩增效果。recA、gyrB、dnaE、dtdS、pntA、pyrC及tnaA基因对应的扩增片段长度分别为773 bp、629 bp、596 bp、497 bp、 470 bp、533 bp、463 bp。
2.3.2管家基因位点的测序及数据分析 将PCR扩增产物委托杭州华大基因公司进行双向测序。测序结果经Chromas软件和DNA Star软件处理后,上传至http://pubmlst副溶血性弧菌数据库(V.parahaemolyticusDatabase Profiles)比对,获得各管家基因位点的等位基因数值,并形成相应的等位基因谱,提交MLST网站,确定副溶血性弧菌O3∶K6血清型菌株的序列分型(Sequence Type,ST),并与数据库中的国内或国际分离菌株做进化关系分析。
3 结 果
3.1管家基因扩增片段结果 采用经过优化的退火温度、分别扩增副溶血性弧菌基因组上的7 个管家基因位点(recA、gyrB、dnaE、dtdS、pntA、pyrC和tnaA基因)目标片段结果,62株菌株均扩增出7 个管家基因片段,扩增产物的核酸浓度及纯度均符合测序要求(图1)。
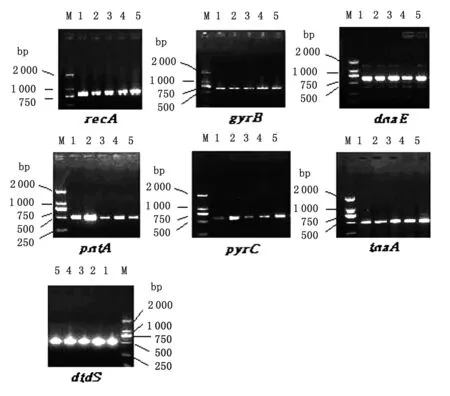
图1 7个管家基因PCR扩增产物琼脂糖凝胶电泳图
Fig.1AgarosegelelectrophoresisofPCRamplificationproductofsevenhousekeepinggenes
1:08-江干-Y-72;2:05-绍-Y-34;3:09-W-367;4:08-宁-Y-4;5:09-安-毒-54;M:DNA marker
3.2副溶血性弧菌MLST分型 将等位基因谱提交MLST网站结果,62株副溶血性弧菌中59株为ST-3型(3,4,19,4,29,4,22)克隆群株,1株菌株MLST序列型为ST-121(3,2,82,52,4,78,66),编号为09-W-114的菌株为新发现的序列型(5,10,34,27,77,49,23),编号为08-江干-Y-75的菌株gyrB基因序列与AP4(Allelic profile)有很大的相似度,仅在第562位点上由T碱基突变为C碱基,其余基因等位基因图谱与ST-3型相同。
3.3副溶血性弧菌系统进化关系 通过在线软件Start2,根据UPGMA法[10]绘制基因系统发生树,分析结果显示:62株副溶血性弧菌及数据库中引用的菌株的序列型分为5个基因群(图2,从上到下依次命名为A群,B群,C群,D群,E群)。
由图2可见,ST-3序列型和ST-121序列型同属C群,但在不同的分枝点上。09-W-114菌株为新发现的序列型,属于D群。ST-3序列型与ST-121序列型的菌株的亲缘关系较近,与09-W-114菌株在进化树上的距离较远。
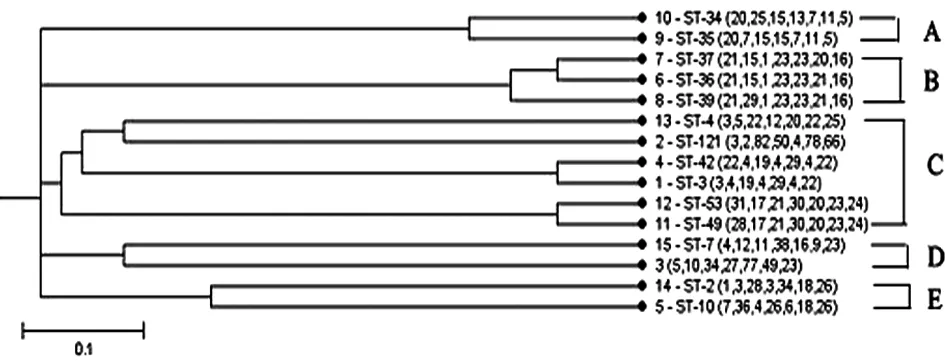
图2 副溶血性弧菌UPGMA系统发生树
4 讨 论
副溶血性弧菌是浙江省主要食源性病原菌,近十年来,由副溶血性弧菌引起的食物中毒事件居细菌性食物中毒的首位,开展副溶血性弧菌遗传进化关系及其群体遗传学特征研究对副溶血性弧菌病的预防和控制具有重要意义。多位点序列分型利用所测定的管家基因的核苷酸序列组合进行编码分型,可有效分析不同时间不同地区临床分离株的遗传相关性,特别是多地域相关性微生物的流行病学、种内微进化、来源追踪和抗生素耐受研究,并且其克服了不同实验室数据重复性及可比性差的缺点,可进行数据共享[7-9]。本次通过对不同来源样品中分离的副溶血性弧菌O3∶K6血清型菌株进行多位点序列分型研究发现:浙江省副溶血性弧菌分离株管家基因的等位基因数仅有3~4个,95.16%(59/62)菌株为ST-3克隆群,从分离对象来看,59株ST-3克隆群副溶血性弧菌29株分离自不同地区门诊腹泻病人,21株分离自15起食物中毒病人,7株分离自食物中毒病人所食用的食物,2株分离自市售食品。表明ST-3克隆群是浙江省高致病性O3∶K6血清型副溶血性弧菌的主要ST克隆群,这与国外Gonzalez-Escalona[10]等的相关研究报导一致。因此,作者认为浙江省在公共卫生监测工作中应重点关注ST-3克隆群副溶血性弧菌,加大对食品,尤其是对海产品的副溶血性弧菌的监测力度,并引导公众形成健康的饮食习惯(食用煮熟的食物),防制由此类病原菌引起的食源性疾病的暴发流行。
参考文献:
[1]Wang Y, Hu QH, Mu J, et al. Etiologic and molecular characteristics ofVibrioparahaemolyticusstrains isolated from diarrheal patients in Shenzhen, in2007-2008[J]. Chin J Epidemiol, 2010, 31(1): 51-55. (in Chinese)
王艺,扈庆华,牟瑾,等.深圳市2007—2008 年腹泻病副溶血性弧菌监测及分子特性分析[J].中华流行病学杂志, 2010,31(1):51-55.
[2]Chowdhury NR, Chakraborty S, Ramamurthy T, et al. Molecular evidence of clonalVibrioparahaemolyticuspandemic strains[J]. Emerg Infect Dis, 2000, 6(6): 631-636. DOI: 10.3201/eid0606.000612
[3]Kubota K, Iwasaki E, Inagaki S, et al. The human health burden of foodborne infections caused byCampylobacter,Salmonella, andVibrioparahaemolyticusin Miyagi Prefecture, Japan[J]. Foodbore Pathog Dis, 2008, 5(5): 641-648. DOI: 10.1089/fpd.2008.0092.
[4]Chiou CS, Hsu SY, Chiu SI, et al.Vibrioparahaemolyticusserovar O3:K6 as cause of unusually high incidence of food-borne disease outbreaks in Taiwan from 1996 to 1999[J]. J Clin Microbiol, 2000, 38(12): 4621-4625.
[5]Liu XM, Chen Y, Wang XY, et al. Foodborne disease outbreaks in China from 1992 to 2001 national foodborne disease surveillance system[J]. J Hyg Res, 2004, 33(6): 725-757. (in Chinese)
刘秀梅,陈艳,王晓英,等.1992-2001年食源性疾病暴发资料分析-国家食源性疾病监测网[J].卫生研究,2004,33:725-757.
[6]Chen N, Sun L, Jiang XG. A summary of food poisoning in Zhenjiang Province in 2007[J]. Zhejiang J Prev Med, 2009, 21(10): 4241-4244. (in Chinese)
陈娜,孙亮,蒋贤根.浙江省2007年度食物中毒情况分析[J].浙江预防医学2009,21(10):4241-4244.
[7]Maiden MC, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenicmicroorganisms[J]. Proc Natl Acad Sci U S A, 1998, 95(6): 3140-3145.
[8]Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer[J]. Mol Biol Evol, 2002, 19(12): 2226-2238.
[9]Liu JH, He D, Yang YQ, et al. Application of multilocus sequence typing method on pathogenic microorganisms typing and identification[J]. Microbiology, 2007, 34 (6): 1188-1191. (in Chinese).
刘金华,贺丹,杨艳秋,等.多位点测序分型技术在病原微生物分型鉴定中的应用[J].微生物学报,2007,34(6):1188-1191.
[10]González-Escalona N, Martinez-Urtaza J, Romero J, et a1. Determination of molecular phylogenetics ofVibrioparahaemolyticusstrains by multilocus sequence typing[J]. J Bacteriol, 2008, 190(8): 2831-2840. DOI: 10.1128/JB.01808-07
猜你喜欢
当代水产(2022年8期)2022-09-20 06:45:40
中老年保健(2022年1期)2022-08-17 06:14:22
食品安全导刊(2021年20期)2021-08-30 06:39:56
食品安全导刊(2021年20期)2021-08-30 06:39:04
中老年保健(2021年6期)2021-08-24 06:54:00
昆明医科大学学报(2021年5期)2021-07-22 07:32:50
当代水产(2019年5期)2019-07-25 07:50:56
现代食品(2016年24期)2016-04-28 08:11:54
广东海洋大学学报(2015年3期)2015-12-22 10:05:26
药学与临床研究(2015年4期)2015-06-05 11:35:52
