
用于MALDI-TOF MS分析的毛细管层析柱制备方法研究
2014-03-26 05:29赵媛媛杨小雁孔英俊康跻耀王明林张贵锋苏志国
生物学杂志 2014年6期
赵媛媛, 杨 帆, 杨小雁, 孔英俊, 康跻耀, 王明林, 张贵锋, 苏志国
(1. 山东农业大学 食品科学与工程学院, 山东 泰安 271018;2. 中国科学院过程工程研究所生化工程国家重点实验室, 北京 100190)
基质辅助激光解吸电离飞行时间质谱(MALDI-TOF MS)是一种基于软电离的生物质谱分析技术,具有高灵敏度、高准确度和高分辨率等优点,在生命科学、医学、药学以及材料科学等领域广泛应用。近年来,随着新型基质及样品预处理技术的研究与应用,MALDI-TOF MS在生物标志物检测、蛋白质测序、微生物鉴定等方面的应用引起广泛关注[1-2]。由于目标物的信号强度受其浓度、溶液中无机盐种类和浓度以及小分子引起的“分子量歧视效应”等因素的影响[3-5],样品预处理方法始终是基于MALDI-TOF MS分析技术应用研究中的热点。
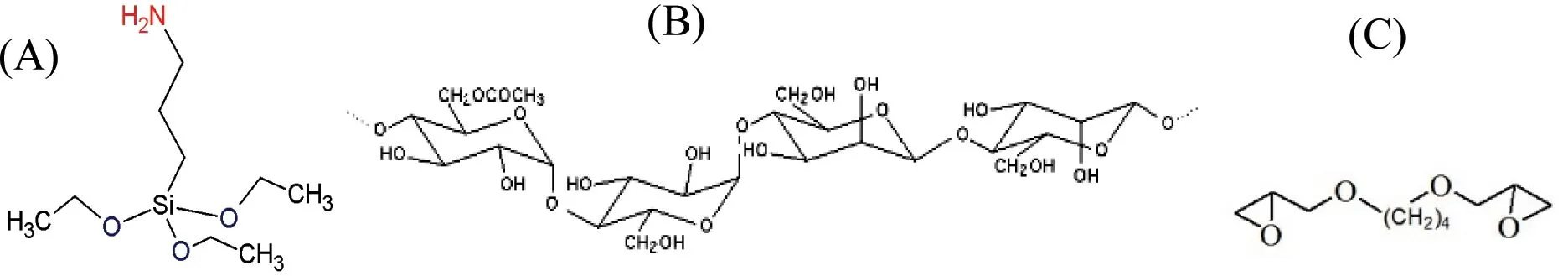
图1 试剂APTES(A)、KGM(B)和BDDE(C)结构式Fig 1 Structure of reagents APTES (A), KGM (B) and BDDE (C)
文献报道的用于MALDI-TOF MS分析的样品预处理技术包括磁性微球技术、固相微萃取技术以及微流控技术等。赵曼等人采用间苯三甲酸铜包被磁性颗粒制备了具有核壳结构的Fe3O4@[Cu3(btc)2]颗粒,对蛋白酶解产物中的多肽具有强吸附性[6],类似方法还包括基于Fe3O4@C@Au和(Fe(3)O(4)@PDA NPs)纳米颗粒等,可用于凝血酶和小分子化合物的质谱分析前的预处理[7-8],基于磁性微球的样品处理过程需经过微球收集、洗涤和点样等过程,会降低样品处理过程的通量。基于微流控的样品预处理技术已有报道,如将微流控芯片与电泳相结合可有效地实现目标物分离,并可将样品转移到MALDI的靶盘上,提高了检测方法的通量[9]。有人利用芯片电泳将目标物分离后进行冷冻处理后直接将芯片置入MALDI靶盘上,但质谱分析中样品厚度对质量精度影响较大,需要对常规的靶盘进行改造[10]。其它样品预处理技术还包括基于Zip-Tip的样品浓缩技术、基于液相色谱分离和自动点样、胶内酶解和提取技术等[11-12],这些样品预处理技术的局限性在于检测限受液相色谱或电泳上样量影响。近年来,基于微层析柱的样品预处理方法研究日益增多[13-15]。毛细管层析柱内可装填离子交换层析介质,用于从蛋白酶解产物中快速富集磷酸化多肽[16]。将亲和配基直接固定到MALDI的靶盘上也可用于目标物快速富集[17],基于亲和层析的样品处理技术可从样品中富集出目标物并用于MALDI-TOF MS分析,如采用金属螯合层析的毛细管柱可实现含磷酸化多肽的快速富集[18-19]等,但传统方法所能处理的样品量低,导致目标物浓度低而难以被检测出。
本研究旨在探索一种毛细管亲和层析柱的制备方法,采用表面氧化法结合氨基硅烷修饰法将氨基引入毛细管内表面,将多糖进行环氧活化后偶联到毛细管,再将蛋白质配基偶联到多糖表面,制成亲和层析柱,利用实际样品对制备的毛细管亲和层析柱有效性进行了验证。
1 材料与方法
1.1 材料与试剂
氨基丙基三乙氧基硅烷(APTES)、芥子酸(SA)、胰蛋白酶和胰蛋白酶抑制剂购自美国Sigma试剂公司,BDDE试剂购自国药集团,色谱纯乙腈、三氟乙酸均购自美国热电公司,魔芋葡甘聚糖(KGM)购自湖北远成赛创公司;其它化学试剂均为市售分析纯试剂。石英毛细管(内径500 μm、690 μm)购自河北锐沣光纤厂。
1.2 主要仪器设备
高效液相色谱仪为美国Agilent公司的HPLC1100,PDA检测器为美国热电公司UV6000检测器(检测池长5 cm),MALDI-TOF MS为德国布鲁克公司的AutoFlex III质谱;X射线光电子能谱(XPS)为美国热电公司的Escalab250Xi设备。
1.3 毛细管层析柱制备方法
毛细管层析柱制备过程,具体过程见图2。

图2 基于毛细管的亲和层析柱制备过程Fig 2 Process for preparation of capillary affinity chromatographic column
1.3.1 毛细管内表面处理方法
截取2 m石英毛细管,用玻璃注射器吸取10 mL超纯水连续洗涤毛细管,注入“Piranha”溶液(体积比为7∶3的95%硫酸和双氧水),90℃恒温处理1 h,用10 mL去离子水冲洗毛细管,使用氮气连续吹脱毛细管内壁20 min。
1.3.2 氨基衍生方法
将5 μmol/L APTES的甲苯溶液注入毛细管,在25℃放置20 min,使用10 mL甲苯洗涤毛细管,使用高纯氮气将毛细管内壁吹干,在烘箱中100℃恒温放置3 h。
1.3.3 KGM活化
按照文献[20]中的方法制备平均分子量为5 kDa、10 kDa和20 kDa的KGM;取20 mg制备的KGM溶于5 mL NaOH溶液(浓度1.5 mol/L,含10 mg/mL硼氢化钠),加入100 μL BDDE, 25℃反应3 h。加入等体积无水乙醇,离心后收集沉淀物,使用80%的乙醇水溶液洗涤沉淀物3次,氮气吹干。
1.3.4 活化KGM偶联
将活化后的KGM溶于0.1 mol/L的NaHCO3-Na2CO3溶液,注入毛细管, 30℃反应2 h,使用10 mL的NaHCO3-Na2CO3缓冲液(0.1 mol/L)连续冲洗毛细管。
1.3.5 蛋白质偶联
将2 mg/mL的胰蛋白酶溶液(0.1 mol/L NaHCO3-Na2CO3缓冲液)注入步骤1.3.4处理后的毛细管,在25℃反应3 h,用10 mL去离子水冲洗毛细管。
1.3.6 环氧基封闭
向毛细管内注入1 mol/L乙醇胺溶液,37℃恒温处理4 h,用5 mL乙酸-乙酸钠缓冲液(0.1 mol/L,pH值4.0,含0.5 mol/L NaCl)连续冲洗毛细管。
1.4 分析方法
1.4.1 总氨基酸含量
将偶联胰蛋白酶的毛细管(1 m)截成长度为3 cm毛细管放置到氨基酸水解瓶内,加入6 mol/L的盐酸,封口后在110℃水解16 h,氮气吹干后使用0.05 mol/L的碳酸氢铵洗涤残留物,吹干后加入50 μL碳酸氢钠溶液(0.05 mol/L)。总氨基酸含量分析方法参照文献[21]。
1.4.2 活化KGM中环氧基含量测定
参照文献[22]所述的方法,其中溶液中的KGM采用加入等体积无水乙醇后离心的方法进行分离,环氧基含量按单位质量的KGM中环氧基物质的量计算(mmol/g)。
1.4.3 XPS分析方法
将毛细管压碎后采用XPS法分析断面元素组成,XPS分析条件:使用带单色器铝靶X射线源,功率为225 W(工作电压15 kV,发射电流15 mA)。污染碳(内标)为284.8 eV;最小能量分辨率0.48 eV(Ag3d5/2),XPS分析区域直径15 μm;数据处理使用Avantage数据系统。
1.4.4 MALDI-TOF MS 分析方法
MALDI-TOF MS的脉冲氮激光(337 nm)离子解析电离源;加速电压20 kV;激光脉冲次数100次/s;质谱扫描范围:m/z 5000~30000;正离子线性模式检测。将SA溶解到70%的乙腈水溶液(v/v,含0.1% TFA),浓度为10 mg/mL;将毛细管层析柱洗脱液直接在靶盘上点样,自然风干加1 μL SA溶液后风干。质谱分析前仪器的质量数利用外标法,使用标准蛋白混合物进行校正。
2 结果与分析
2.1 氨基硅烷修饰前后毛细管的XPS分析结果
石英毛细管主要成分是SiO2,为了将羟基引入到毛细管内表面,实验首先采用Piranha溶液对毛细管内壁进行了处理,在浓H2SO4和H2O2强氧化条件将羟基引入石英毛细管内表面。APTES中的乙氧基可与羟基反应,为了将氨基偶联到毛细管内表面,实验采用APTES的甲苯溶液对毛细管的内表面进行了修饰。采用XPS对APTES处理前后的毛细管(690 μm)内表面元素组成进行了分析。
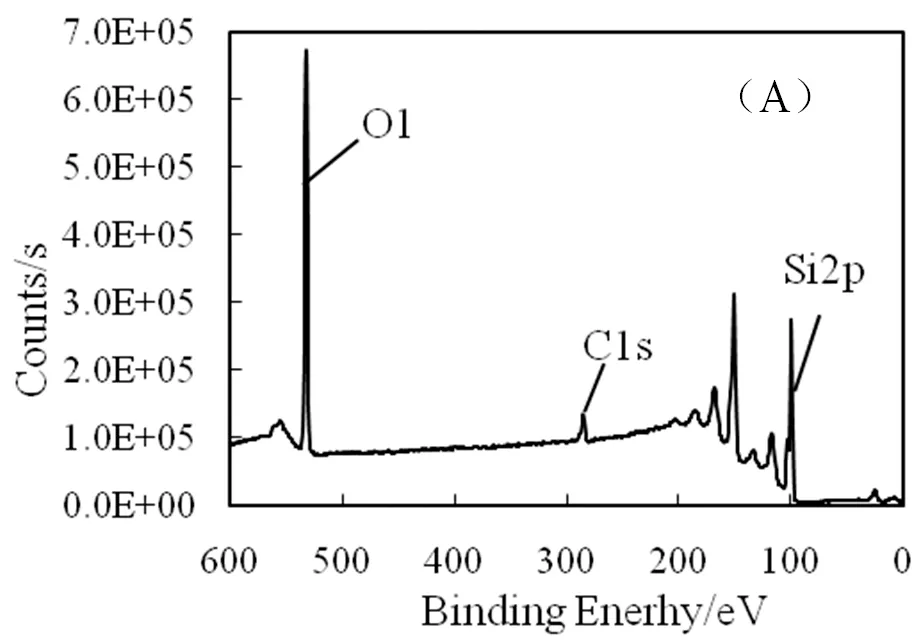
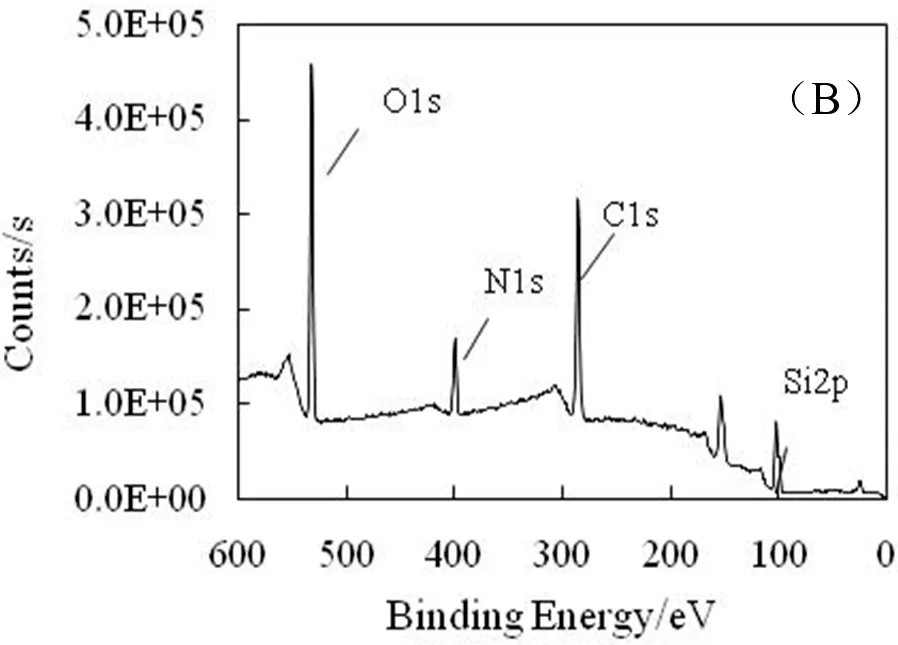
图3 APTES修饰前(A)和修饰后(B)的石英毛细管表面XPS分析结果
图3是APTES修饰前后毛细管内表面XPS图谱,修饰后毛细管表面出现结合能为399.4 eV的信号峰,与氨基中N1S结合能对应;同时,毛细管表面出现结合能为284.8 eV的信号峰,与APTES中的C-C键对应, C1s信号峰也出现在285.8 eV,与APTES中C-N键对应,表明修饰后的毛细管上出现C-C键与C-N键。修饰前后的XPS图谱表明氧化处理的毛细管经APTES进一步修饰后可将-NH2成功地引入到毛细管内表面。APTES修饰后的毛细管上N和C的元素组成分别为4.1%和25.3%,比例接近1∶6,表明在实验条件下每个APTES分子中参与反应的乙氧基数量并不相同。实验利用类似的方法研究了APTES浓度和修饰时间对毛细管内表面N元素含量的影响,结果表明在APTES浓度超过0.005 mol/L浓度条件,反应时间超过20 min后对N含量影响并不显著,APTES浓度低于0.001 mol/L条件下在10~40 min范围内修饰表面上N含量与反应时间相关。
2.2 活化KGM的环氧基含量分析
经BDDE活化后的KGM分子中环氧基团含量是影响蛋白质偶联量重要因素,主要受活化反应的时间和活化温度影响,实验前期研究了KGM微球表面上环氧配基含量与温度和时间之间的关系[22],由于在反应温度超过50℃条件下受水解反应影响,环氧基团含量会出现先升高后降低的趋势,50℃温度较高,环氧基水解反应快,环氧开环使环氧基含量迅速降低,在30℃或更低温度条件下环氧基开环反应的速率较低,环氧基含量达到峰值后缓慢降低。由于KGM微球与未交联的KGM活化方法存在一定差异,实验重点考察了25℃条件下反应时间对KGM分子中环氧基团含量的影响。如图4所示,在反应初始阶段环氧基团与KGM比例逐渐升高,反应在15 h后环氧基含量增长缓慢。为了考察环氧反应对后续蛋白偶联量的影响,实验分别制备了环氧基含量为1.1、1.9和2.8 mmol/g的活化KGM。
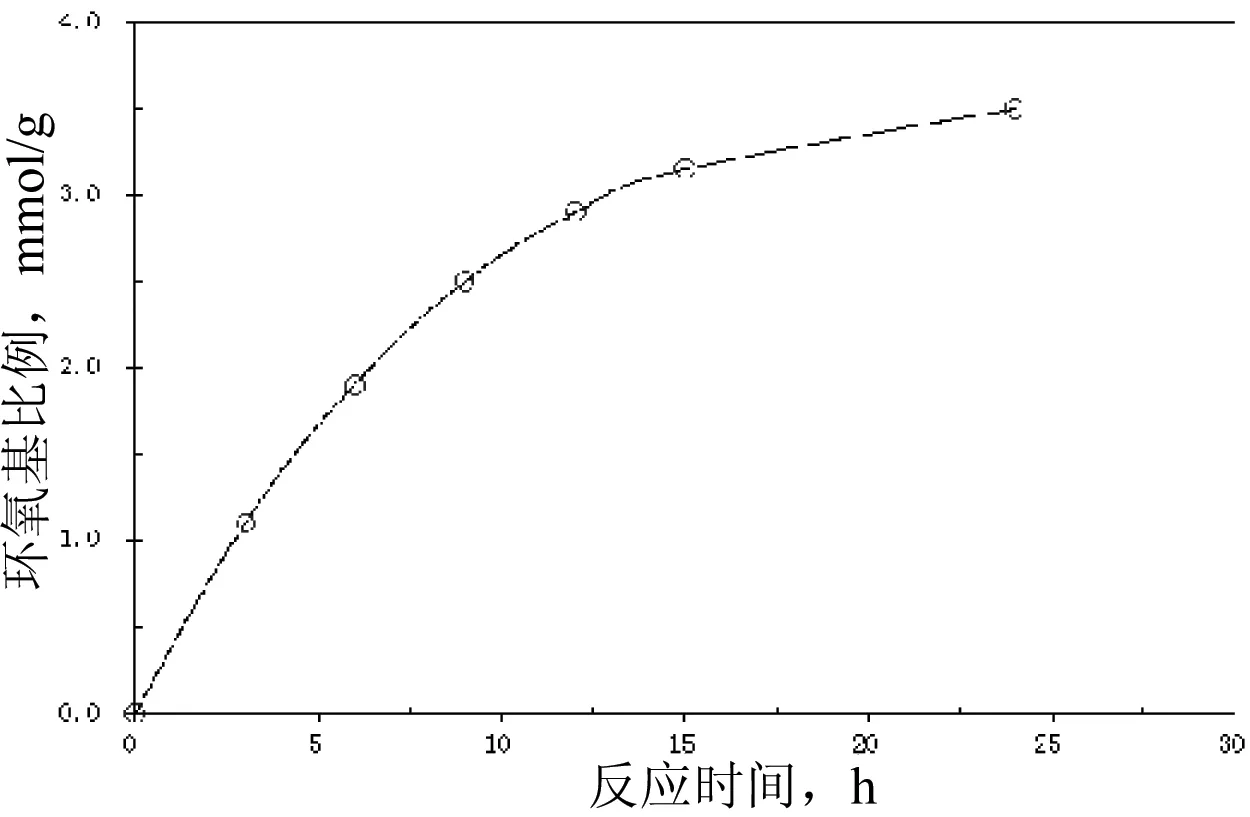
图4 反应时间对环氧基团与KGM(10 kDa)比例的影响
2.3 蛋白偶联量分析
亲和层析过程受毛细管表面上蛋白质配基的偶联量影响。由于直接分析毛细管内蛋白质含量较为困难,实验采用测定总氨基酸的方法研究胰蛋白酶偶联量相对变化,并进一步考察了KGM分子量对偶联量的影响。在环氧基含量为2.8 mmol/g条件下,采用20 kDa的活化KGM偶联后毛细管内胰蛋白酶的偶联量为54 μg/m,当KGM平均分子量为10 kDa和5 kDa时,毛细管内胰蛋白酶的偶联量分别为33 μg/m和27 μg/m,表明KGM分子量越高则多糖链越长,可偶联的蛋白质分子数越多(图5)。进一步地研究结果表明KGM(20 kDa)上环氧基团含量越高,则单位长度的毛细管上可偶联的胰蛋白酶量越多,但当超过一定极限值后,胰蛋白酶偶联量不再受环氧基团含量影响,其可能原因在于胰蛋白酶分子半径较大,偶联的蛋白质数量受蛋白质的空间位阻影响。

图5 KGM分子量对毛细管内胰蛋白酶偶联量的影响

图6 含胰蛋白酶溶液(A)和平均分子量为5 kDa(B)和20 kDa KGM(C)偶联胰蛋白酶层析柱处理后样品MALDI-TOF MS图谱
2.4 毛细管层析柱性能评价
实验首先采用BSA为模型蛋白(0.1 mg/mL, 0.05 mol/L NH4HCO3)上样至毛细管层析柱,在37℃条件下恒温处理12 h后,将柱内溶液直接点样到MALDI靶盘上,可检测出胰蛋白酶降解BSA后形成的多肽,证明了胰蛋白酶被成功偶联在毛细管层析柱内。实验进一步采用胰蛋白酶抑制剂(10 μg/mL,0.05 mol/L Tris-HCl,pH值7.0)对固定化胰蛋白酶的毛细管层析柱(6 cm长,内径500 μm)分离性能进行了验证,图6A是原液的MALDI-TOF MS图谱,图6B和6C分别是采用平均分子量为5 kDa和20 kDa KGM偶联胰蛋白酶层析柱,各上样500 μL后洗脱液直接点样后的MALDI-TOF MS图谱。从图6中离子种类和相对丰度可知,原样中未检测出胰蛋白酶抑制剂离子,经毛细管层析柱处理后样品中检测出m/z19988.4的离子,以环氧活化后的20 kDa KGM为链接剂偶联胰蛋白酶后的层析柱的亲和分离效果明显优于5 kDa KGM,可见,提高KGM的分子量有助于增加层析柱上胰蛋白酶的偶联量。
本研究表明采用所制备的毛细管亲和层析柱可有效分离出胰蛋白酶抑制剂,基于毛细管亲和层析柱的样品预处理方法具有可行性。
参考文献:
[1]Rodrigo M A M, Zitka O, Krizkova S, et al. MALDI-TOF MS as evolving cancer diagnostic tool: a review[J]. J Pharm Biomed Anal, 2014, 95: 245-255.
[2]Sandrin T R, Goldstein J E, Schumaker S. MALDI TOF MS profiling of bacteria at the strain level: a review[J]. Mass Spectr Rev, 2013, 32(3): 188-217.
[3]Fuchs B, Schiller J. MALDI-TOF MS: much more than only protein analysis[J]. J Glycomics Lipidomics, 2013, 3(1): E113.
[4]Weidmann S, Mikutis G, Barylyuk K, et al. Mass discrimination in high-mass MALDI-MS[J]. J Am Soc Mass Spectr, 2013, 24(9): 1396-1404.
[5]Knochenmuss R. MALDI and related methods: a solved problem or still a mystery?[J] Mass Spectrom, 2013, 2: S0006.
[6]Zhao M, Deng C H, Zhang X M, et al. Facile synthesis of magnetic metal organic frameworks for the enrichment of low-abundance peptides for MALDI-TOF MS analysis[J]. Proteomics, 2013, 13(23-24): 3387-3392.
[7]Zhang X Y, Zhu S C, Deng C H, et al. Highly sensitive thrombin detection by matrix assisted laser desorption ionization-time of flight mass spectrometry with aptamer functionalized core shell Fe3O4@C@Au magnetic microspheres[J]. Talanta, 2012, 88(15): 295-302.
[8]Ma Y R, Zhang X L, Zeng T, et al. Polydopamine-coated magnetic nanoparticles for enrichment and direct detection of small molecule pollutants coupled with MALDI-TOF-MS[J]. ACS Appl Mater Interface. 2013, 5(3): 1024-30.
[9]Kitagawa F, Otsuka K. Recent progress in microchip electrophoresis mass spectrometry[J]. J Pharm Biomed Anal, 2011, 55(4): 668-678.
[10]Nie L, Xu G B, Wang X Y, et al. Coupling microchip electrophoresis with MALDI-TOF-MS based on a freezing technique[J]. Chinese Chem Lett, 2013, 24: 491-493.
[11]Jakoby T, Berg B H J, Tholey A. Quantitative protease cleavage site profiling using tandem-mass-tag labeling and LC MALDI-TOF/TOF MS/MS analysis[J]. J Proteome Res, 2012, 11 (3): 1812-1820.
[12]Almeida A, Ferreira J A, Teixeira F, et al. Challenging the limits of detection of sialylated Thomsen Friedenreich antigens by in-gel deglycosylation and nano-LC-MALDI-TOF-MS[J]. Electrophoresis,2013, 34(16): 2337-2341.
[13]Horka M, Kubesova A, Salplachta J, et al. Capillary and gel electromigration techniques and MALDI-TOF MS-Suitable tools for identification of filamentous fungi[J]. Anal Chim Acta, 2012, 716(24): 155-162.
[14]Jensen P H, Mysling S, Hojrup P, et al. Glycopeptide enrichment for MALDI-TOF mass spectrometry analysis by hydrophilic interaction liquid chromatography solid phase extraction[J]. Method Mol Biol, 2013, 951: 131-144.
[15]Liu J, Wang F J, Lin H, et al. Monolithic capillary column based glycoproteomic reactor for high-sensitive analysis of N-glycoproteome[J]. Anal Chem, 2013, 85 (5): 2847-2852.
[16]Dong M M, Wu M H, Wang F J,et al. Coupling strong anion-exchange monolithic capillary with MALDI-TOF MS for sensitive detection of phosphopeptides in protein digest[J]. Anal Chem, 2010, 82 (7): 2907-2915.
[17]Ahmad-Tajudina A, Adler B, Ekstrom S, et al. MALDI-target integrated platform for affinity-captured protein digestion[J]. Anal Chim Acta, 2014, 807: 1-8.
[18]Dai L, Preston R, Bacica M,et al. Development of a potential high-throughput workflow to characterize sites of bioconjugation by immuno-affinity capture coupled to MALDI-TOF mass spectrometry[J]. Bioconjugate Chem., 2013, 24 (1): 53-62.
[19]Zhang L Y, Wang H, Liang Z, et al. Facile preparation of monolithic immobilized metal affinity chromatography capillary columns for selective enrichment of phosphopeptides[J]. J Sep Sci, 2011, 34(16/17): 2122-2130.
[20]姚 雪,罗学刚,韩本超. 魔芋葡甘聚糖酶解过程及各种不同分子量魔芋葡甘低聚糖制备条件的研究[J]. 食品工业科技,2011, 32(9): 97-101.
[21]王 棘,战祥友,腾艳坤,等. DNFB柱前衍生化RP-HPLC法测定氨肽素的氨基酸含量[J]. 沈阳药科大学学报,2003, 20(6): 428-430.
[22]康跻耀, 张 宁, 周炜清,等. 基于魔芋葡甘聚糖微球的胶原覆层型微载体的制备研究[J]. 中国生物工程杂志,2013,33(5): 44-49.
猜你喜欢
大电机技术(2022年5期)2022-11-17
云南化工(2020年11期)2021-01-14
世界农药(2019年4期)2019-12-30
心肺血管病杂志(2019年1期)2019-04-22
材料科学与工程学报(2016年1期)2017-01-15
当代化工研究(2016年9期)2016-03-20
杂草学报(2015年2期)2016-01-04
中药与临床(2015年5期)2015-12-17
中国塑料(2015年8期)2015-10-14
食品工业科技(2014年5期)2014-03-11
