
Activation of Nitromethane to Cyanide by a Mononuclear Cu(II) Complex①
2014-03-25 02:35WUNnNnCHENChngNengHUANGDeGung
结构化学 2014年11期
WU Nn-Nn CHEN Chng-Neng HUANG De-Gung②
a (State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China)
b (University of Chinese Academy of Sciences, Beijing 100039, China)
1 INTRODUCTION
The research interest of small molecule activation increased with the growth of world economy and thereafter energy & environmental problems. Activation led to utilization of the compound as a renewable energy resource in the formation of useful organic compounds and/or function groups, ideally in catalytically efficient systems. Utilization of organic solvents by chemical activation and catalytic transformation has received much attention from the viewpoints of thermodynamic stability of products,vast capacity of raw material and potential economic feasibility. Nitromethane is commonly used as a clean solvent in industrial applications, as a fuel in internal combustion engines, and as a reaction medium for organic synthesis, in which CH3NO2was mostly employed as a one carbon source[1,2]. Its acidity allowed it to undergo deprotonation reactions to form a variety of carbon-containing species.
The thermal decomposition of nitromethane under high pressure and high temperature has been investigated since 1950s[3-5]. The main products of pyrolysis of nitromethane were nitric oxide,hydrogen cyanide, nitrogen dioxide, formaldehyde and so forth[6-7]. Limão-Vieira et al. have studied the ion-pair formation in gaseous nitromethane by electron transfer as well as the products of collisions between fast potassium atoms and nitromethane molecules. The negative ions formed in such collisions contained NO-, CH2NO2-, OH-, CNO-and CN-[8]. In this paper, we report a new experimental investigation of CH3NO2activation reaction by a mononuclear CuIIcomplex to form CN-under mild conditions (Scheme 1). This work might offer an indication that highly functioned coordination complexes catalysts for solvent activation could be constructed.
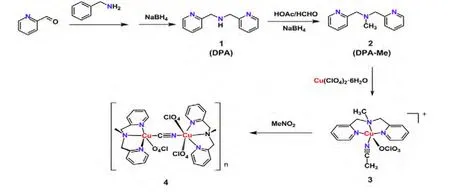
Scheme 1. Route for the activation of CH3NO2 by formation of a dinuclear complex {[Cu(DPA-Me)]2(CN)(ClO4)3·CH3NO2}n (4)
2 EXPERIMENTAL
2.1 General procedures
Unless otherwise stated, all reactions were carried out under a dinitrogen atmosphere using Schlenk techniques. Methanol was dried over 3 Å molecular sieves before use. Other solvents were of reagent grade and used as received. IR spectra were obtained on a Perkin-Elmer Spectrum One FT-IR spectrometer with KBr pellets. UV-Vis spectra were measured on a Perkin-Elmer Lambda 950 spectrometer.Elemental analysis of C, H and N was carried out on a Vario EL III CHNOS elemental analyzer. NMR spectra were recorded using a Bruker Avance III(400 MHz) spectrometer. Chemical shifts were given in ppm relative to TMS (1H, 0.0 ppm).
2.2 Synthesis of 2,2΄-dipicolylamine (1)
The reported method[9]was modified for the synthesis of this precursor. A mixture of 2-pyridinecarboxaldehyde (2.14 g, 20.0 mmol) and 2-aminomethylpyridine (2.16 g, 20.0 mmol) in methanol(100 mL) was stirred for 12 h at room temperature.Sodium borohydride (1.89 g, 50.0 mmol) was added in small portions at 0 ℃ and the resulting mixture was heated to reflux for 3 h. Solvent was rotated off and 2M NaOH (aq) was added until the pH was ca.14. The aqueous solution was extracted with CH2Cl2(3 × 80 mL) and the organic layers were combined and dried over anhydrous Na2SO4. Solvent was removed and the sticky residue was purified on a silica column eluted with CH2Cl2/MeOH/NH3·H2O(70/5/1, v/v/v) to give product 1 as a yellow oil (2.79 g, 70% based on Py-CH2-NH2).1H NMR (400 MHz,CDCl3): δ 8.56 (d, 2H, J = 4.5 Hz, PyH), 7.64 (t, 2H,J = 7.7, 1.7 Hz, PyH), 7.36 (d, 2H, J = 7.8 Hz, PyH),7.16 (t, 2H, J = 7.2, 5.1 Hz, PyH), 3.95 (d, 4H,CH2Py), 2.65 (s, 1H, br, NH).13C NMR (101 MHz,CDCl3): δ 159.72, 149.24, 136.49, 122.25, 121.91,54.78.
2.3 Synthesis of N-methyl-2,2΄-dipicolylamine (2)[10]
To a solution of 1 (1.2 g, 6.02 mmol) in acetonitrile (100 mL) was added acetic acid (15 mL) and an aqueous solution of formaldehyde (8 mL, 37%). The mixture was stirred for 1 h, and sodium borohydride(0.567 g, 15.0 mmol) was slowly added at 0 ℃.Stirring was continued for 36 h at room temperature.All the volatiles were removed and 2M NaOH (aq)was added until the pH was ca. 14, followed by extraction with CH2Cl2(3 × 60 mL). The combined organic layers were washed with water (80 mL),dried over Na2SO4, and filtered. Evaporation of solvent gave product 2 as a light yellow oil (1.11 g,87% based on ligand 1).1H NMR (400 MHz,CDCl3): δ 8.55 (m, 2H, PyH), 7.66 (t, J = 7.7, 1.7,2H, PyH), 7.52 (d, J = 7.8, 2H, PyH), 7.16 (t, J = 6.8,5.4, 2H, PyH), 3.78 (s, 4H, CH2Py), 2.33 (s, 3H,CH3).13C NMR (101 MHz, CDCl3): δ 159.48,149.05, 135.48, 123.03, 121.98, 63.58, 42.83.
2.4 Synthesis of[(DPA-Me)Cu(MeCN)(ClO4)](ClO4) (3)
A solution of Cu(ClO4)2·6H2O (0.204 g, 0.55 mmol) in methanol (5 mL) was added to a solution of 2 (0.117 g, 0.55 mmol) in methanol (25 mL). The mixture was stirred for 3 h at room temperature and solvent was rotated off to leave a bluish-green sticky residue. The residue was dissolved in acetonitrile(0.5 mL) and the solution was added dropwise to CH2Cl2(5 mL) to deposit some blue precipitate. The precipitate was collected, dissolved in acetonitrile (2 mL) and diffused with diethyl ether to yield product as some blue crystals 3 in several days (190 mg,67% based on ligand 2). Anal. Calcd. (%) for C15H18Cl2CuN4O8: C, 34.86; H, 3.51; N, 10.84.Found (%): C, 36.60; H, 3.52; N, 10.09. IR (KBr,cm-1): 2933 v(CH3)(w), 1614 v(py-H)(s), 1490 v(py-H)(m), 1450 v(py-H)(s), 1101 v(ClO4)(vs, br).
2.5 Synthesis of{[Cu(DPA-Me)]2(CN)(ClO4)3·CH3NO2}n (4)
Compound 3 (10.3 mg, 0.02 mmol) was dissolved in nitromethane (2 mL) to make a clear blue solution.Diffusion of diethyl ether into this solution deposited a mixture of purple and blue crystals. The mixed crystals were collected and recrystallized in CH3NO2/Et2O, and the process was repeated until no blue crystal was found in the product, yield 1.9 mg,20% based on compound 3. Anal. Calcd. (%) for C28H33Cl3Cu2N8O14: C, 35.81; H, 3.54; N, 11.93.Found (%): C, 35.90; H, 3.43; N, 11.82. IR (KBr,cm-1): 2930 v(CH3)(w), 2199 v(CN)(m), 1612 v(py-H)(s),1485 v(py-H)(m), 1450 v(py-H)(s), 1092 v(ClO4)(vs, br).
2.6 X-ray crystallography
Diffraction quality crystals for 3 and 4 were obtained from the solvents of MeCN/Et2O and CH3NO2/Et2O. A blue single crystal of 3 (0.50mm ×0.40mm× 0.12mm) and a purple single crystal of 4(0.20mm × 0.12mm × 0.02mm) were selected for data collection. The X-ray diffraction data of these two crystals were collected on an Oxford Diffraction Supernova dual diffractometer with MoKα radiation(λ = 0.71073 Å) and CuKα radiation (λ = 1.54184 Å)at 100 K, respectively.
The data reduction and cell refinement were processed using CrysAlisPro software[11]. The structure was solved by direct methods with SHELXS-97 and refined by full-matrix least-squares fitting on F2by SHELXL-97[12,13]. Two perchlorate anions and one nitromethane solvent molecule in 4 were treated as disordered models and the temperature factors of those atoms were restrained by DELU/SIMU to achieve an anisotropic average. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were added geometrically and refined anisotropically. For 3, a total of 17496 reflections (3875 unique) were measured in the range of 2.81≤θ≤26.00°, of which 3175 were observed with I > 2σ(I). The final R = 0.0350, wR =0.0792 (w = 1/[σ2(Fo2) + (0.0344P)2+ 0.8107P],where P = (Fo2+ 2Fc2)/3), (Δρ)max= 0.451, (Δρ)min=–0.481 e·Å-3, (Δ/σ)max= 0.001 and S = 1.062. For 4,37374 total reflections (6978 unique) were measured in the range of 4.04≤θ≤69.99°, of which 6414 were observed with I > 2σ(I). The final R = 0.0485,wR = 0.1246 (w = 1/[σ2(Fo2) + (0.0630P)2+8.4659P], where P = (Fo2+ 2Fc2)/3), (Δρ)max= 1.468,(Δρ)min= –0.724 e·Å-3, (Δ/σ)max= 0.001 and S =1.069. The selected bond lengths and bond angles are listed in Table 1.
3 RESULTS AND DISCUSSION
3.1 Ligand synthesis and nitromethane activation
The intermediate 1 (DPA) was easily made by a condensation reaction and afterwards reduction with NaBH4in yield 70%. Reaction of DPA with HCHO under acid condition gave the desired ligand 2 in high yield (87%). Both 1 and 2 were characterized by1H and13C NMR spectroscopy.
Ligand DPA-Me and Cu(ClO4)2·6H2O were mixed in MeOH to generate the mononuclear Cu(II)complex 3 in 67% yield, in which the copper center was five-coordinated, leaving one axial position open for attack. Furthermore, the two coordinated groups, MeCN solvate molecule and perchlorate anion, were both good leaving groups in dynamics so that the opportunity of attacking by small molecules increased in geometry. Herein the open coordination mode and the basic reaction condition might be attributed to the driving force for the activation of CH3NO2molecule in solution. Recrystallization of 3 in CH3NO2several times in air led to a change of colour from blue to purple slowly, from which a kind of purple crystal was isolated and identified by X-ray crystallography as a [Cu(II)-X≡X’-Cu(II)]3+species. The bond length (X–X:1.152(4) Å) and the bond angles (Cu–X–X’175.3(3)º and X–X’–Cu 178.3(3)º) indicated the existence of a triple bond of X≡X’, which was consistent with the signal shift found at 2199 cm-1for complex 4 in the IR spectroscopy. Together with the assignment of charge of X≡X’ as -1 in crystal structure and the fitting of temperature factors of X&X’ as C, N or O, the structure of[Cu(II)–C≡N–Cu(II)]3+was finally confirmed. Thus,it was certain that the CH3NO2could be activated by mononuclear Cu(II) compound 3 slowly to give the homodinuclear cyanide-bridged Cu(II) complex 4.The reaction mechanism was still not clear.
3.2 Description of the structures of compounds 3 and 4
The molecular structure of 3 is shown in Fig. 1.The structure of 3 contained a mononuclear{[Cu(DPA-Me)(MeCN)(ClO4)]+unit and one isolated perchlorate counteranion outside. The central Cu2+ion presented strongly distorted square pyramidal geometry with the equatorial plane defined by three nitrogen atoms from ligand DPA-Me and one nitrogen atom from the acetonitrile molecule. The Cu2+ion was 0.132 Å out of this N4plane. The axially elongated position of this pyramid was occupied by an oxygen atom from the perchlorate anion with the Cu–O bond length of 2.423(2) Å. The other perchlorate anion possessed the sixth coordination position through a weak intermolecular interaction between Cu(1) and O(6) as 2.597(1) Å.The selected bond lengths and bond angles of 3 are given in Table 1.
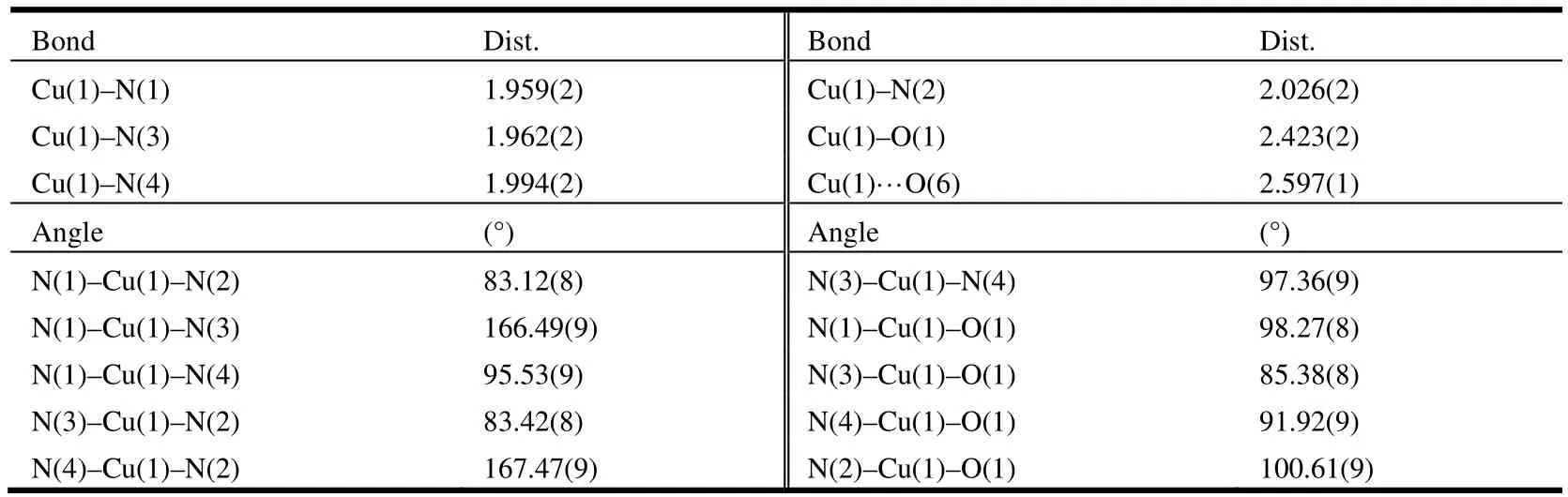
Table 1. Selected Bond Lengths (Å) and Bond Angles (º) for Compound 3
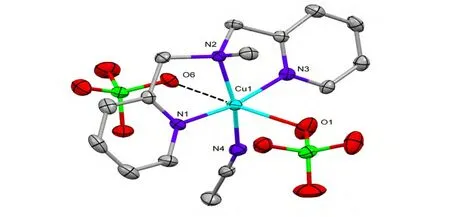
Fig. 1. Molecular structure of compound 3. All hydrogen atoms were omitted for clarity
The unit cell of 4 shows a cyanide-bridged{[Cu(DPA-Me)]2(CN)(ClO4)3} dimmer and one isolated nitromethane solvate molecule (Fig. 2).Either of Cu(II) centers was six-coordinated showing strongly distorted octahedral geometry with the equatorial plane defined by three nitrogen atoms from ligand DPA-Me and one C/N atom from cyanide group. The axially elongated positions were occupied by two oxygen atoms from the perchlorate anions with Cu–O bond lengths being 2.591(1) and 2.629(7) Å for Cu(1), and 2.460(3) Å and 2.537(8)Å for Cu(2) (Fig. 2). The definition of the specific atoms for CN-group was challenged, since the electron densities of C and N atoms in X-ray crystallography were too close to be distinguished. In this structure, two sets of cyanide atoms (C&N or N&C) were refined respectively, aiming to find a better model for the diffraction data, and finally the refinement of C&N set was found giving a better result compared to the N&C set, either in the presentation of atomic temperature factor or in the final given g values. Thus, the atom coordinated to the Cu(1) was set as C(27) and the other one to Cu(2)as N(7). The bond lengths and bond angles of[Cu-C≡N-Cu]3+dimmer were found close to the reported [M-C≡N-M’] units formed in similar coordination modes[10,14-16]. In this case, the packing diagram of 4 showed a one-dimensional chain along the c-axis linked by one {[Cu (DPAMe)]2(CN)(ClO4)3} unit and one perchlorate anion at regular intervals (Fig. 3). The selected bond lengths and bond angles of 4 are presented in Table 2.
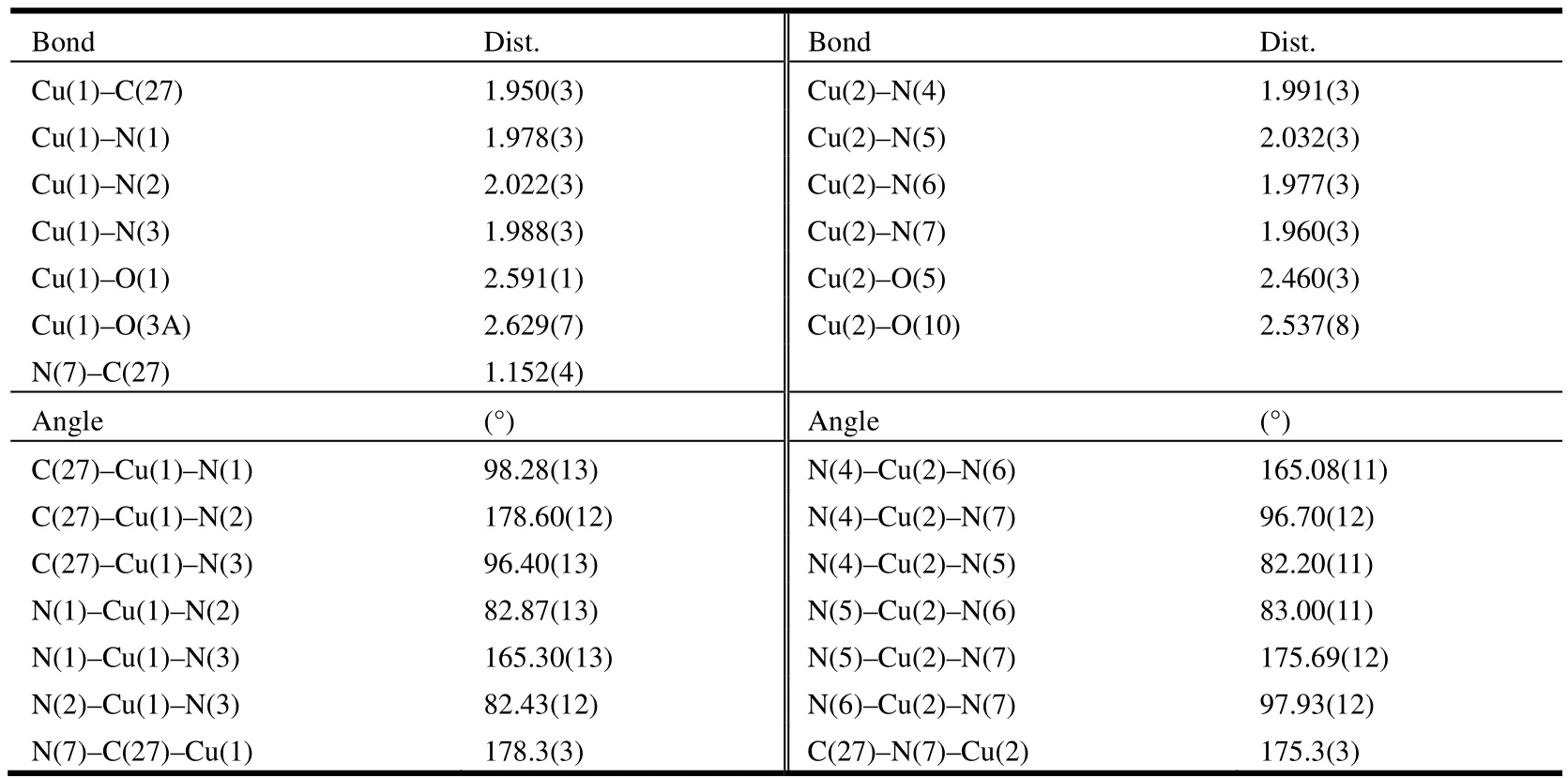
Table 2. Selected Bond Lengths (Å) and Bond Angles (º) for Compound 4
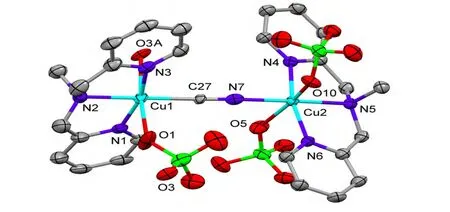
Fig. 2. Molecular structure of compound 4. Solvate molecules and all hydrogen atoms were omitted for clarity
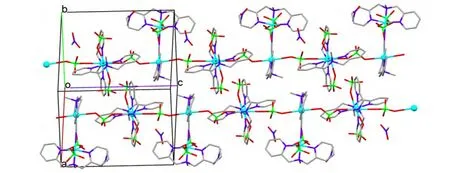
Fig. 3. Packing diagram of 4 showing one-dimensional chain along the c-axis
4 CONCLUSION
In summary, mononuclear {[Cu(DPAMe)(MeCN)(ClO4)](ClO4) was prepared easily and used for the activation of CH3NO2to CN-by the formation of a dinuclear single cyanide bridged{[Cu(DPA-Me)]2(CN)(ClO4)3} complex under mild conditions. Both the starting material and the product complexes were characterized by X-ray crystallography, IR spectroscopy and elemental analysis.
(1) Markofsky, S. B. "Nitro compounds, aliphatic". In Ullmann's Encyclopedia of Industrial Chemistry 2000, Weinheim: Wiley-VCH.
(2) Balamurugan, R.; Manojveer, S. Gold/copper-catalyzed activation of the aci-form of nitromethane in the synthesis of methylene-bridged bis-1,3-dicarbonyl compounds. Chem. Commun. 2011, 47, 11143–11145.
(3) Cottrell, T. L.; Graham, T. E.; Reld, T. J. The thermal decomposition of nitromethane. Trans. Faraday Soc. 1951, 47, 584–590.
(4) Makovky, A.; Gruenwald, T. B. The thermal decomposition of nitromethane under high pressure. Trans. Faraday Soc. 1959, 55, 952–958.
(5) Crawforth, C. G.; Waddington, D. J. Reactions of nitroalkanes in the gas phase. Part 1.—pyrolysis of nitromethane. Trans. Faraday Soc. 1969, 65,1334–1349.
(6) Hu, W. F.; He, T. J.; Chen, D. M.; Liu, F. C. Theoretical study of the CH3NO2unimolecular decomposition potential energy surface J. Phys. Chem. A 2002, 106, 7294–7303.
(7) Kuznetsov, N. M.; Petrov, Y. P.; Turetskii, S. V. Analysis of nitromethane thermal decomposition at low temperatures. Kinetics and Catalysis 2013,54, 131–138.
(8) Antunes, R.; Almeida, D.; Martins, G.; Mason, N. J.; Garcia, G.; Maneira, M. J. P.; Nunes, Y.; Limão-Vieira, P. Negative ion formation in potassium-nitromethane collisions. Phys. Chem. Chem. Phys. 2010, 12, 12513–12519.
(9) Wong, Y. L.; Mak, C. Y.; Kwan, H. S.; Lee, H. K. Mononuclear iron(III) complexes supported by tripodal N3O ligands: synthesis, structure and reactivity towards DNA cleavage. Inorg. Chim. Acta 2010, 363, 1246–1253.
(10) Huang, D.; Holm, R. H. Reactions of the terminal Ni-OH group in substitution and electrophilic reactions with carbon dioxide and other substrates:structural definition of binding modes in an intramolecular NiII -FeII bridged site. J. Am. Chem. Soc. 2010, 132, 4693–4701.
(11) CrysAlisPro, Oxford Diffraction (Poland) 2010.
(12) Sheldrick, G. M. SHELXS-97, Program for the Solution of Crystal Structure. University of Göttingen, Germany 1997.
(13) Sheldrick, G. M. SHELXL-97, Program for the Refinement of Crystal Structure. University of Göttingen, Germany 1997.
(14) Huang, D.; Deng, L.; Sun, J.; Holm, R. H. Cleavage of Ni-(μ2-S)-Ni bridges in dinuclear nickel(II) dithiolate pincercomplexes and related reactions.Inorg. Chem. 2009, 48, 6159–6166.
(15) Rodríguez-Fortea, A.; Alemany, P.; Alvarez, S.; Ruiz, E. Exchange coupling in cyano-bridged homodinuclear Cu(II) and Ni(II) complexes: synthesis,structure, magnetism, and density functional theoretical study. Inorg. Chem. 2001, 40, 5868–5877.
(16) Flay, M. L.; Vaherenkamp, H. Cyanide-bridge oligonuclear complexes containing Ni-CN-Cu and Pt-CN-Cu. Eur. J. Inorg. Chem. 2003, 9, 1719–1726.

- 结构化学的其它文章
- Synthesis and Crystal Structure of(Z)-2-Methyl-5,6-dihydrobenzo[d]thiazol-7(4H)-one O-Prop-2-yn-1-yl Oxime Derivatives①
- Synthesis and Crystal Structure of Cobalt(II) and Copper(II) Complexes Involving L-Aamino Alcohols
- Synthesis, Crystal Structure and Photoluminescence of a Three-coordinate Ag(I) Complex①
- Synthesis, Crystal Structure and Antimicrobial Activity of Ethyl 2-(1-cyclohexyl-4-phenyl-1H-1,2,3-triazol-5-yl)-2-oxoacetate①
- Synthesis, X-ray Crystallographic Analysis and Bioactivities of α-Aminophosphonates Featuring Pyrazole and Fluorine Moieties①
- Syntheses and Structural Characterizations of a Series of Capped Keggin Derivatives①