
Synthesis, Structure and Property of One Bis(benzimidazolin-2-ylidene) Arene-mercury(II) Complex①
2014-03-25 02:35LIYouGuiHANLeiMAGangZHUChengFeng
结构化学 2014年11期
LI You-Gui HAN Lei MA Gang ZHU Cheng-Feng
(School of Chemical Engineering, Hefei University of Technology, Hefei 230009, China)
1 INTRODUCTION
Coordination-driven self-assembly provides efficient opportunities to design and prepare ordered arrays of molecules with unique structures and functions, and has led to significant progress in the supramolecular coordination complexes (SCCs).SCCs, as one of the branches of modern coordination chemistry, are attracting increasing attention not only because of their intriguing structures but also due to their potential promising applications in catalysis, adsorption, separation, optics, and so on[1-10]. Unlike traditional inorganic materials, these materials containing organic units feature structural diversity and amenability to be designed with desired functionalities at the molecular level[11-14].The triazloe, imidazole and benzimidazole-containing ligands are particularly appealing in the construction of coordination complexes for their high affinity to metals, the versatile coordination modes and the variable conformations[15-17]. Herein, we synthesize a pyridyl- and benzimidazolyl-difunctionalized ligand and employ it to assemble with mercury(II) ions to afford a supramolecular coordination complex, and its structure and photoluminescence were studied.
2 EXPERIMENTAL
2.1 General procedure.
All reactions were carried out under a dry nitrogen atmosphere using standard Schlenk techniques, unless stated otherwise. The solvents were purified by standard methods. The melting point was determined with a WRS-1A digital melting point apparatus.1H NMR and13C NMR spectra were recorded at 600 and 100 MHz, respectively on a Bruker 400 spectrometer. Chemical shifts, δ, are reported in ppm relative to the internal standard TMS. J values are given in Hz. Elemental analyses were measured using a Yanaco MT-3 elemental analyzer. UV-vis spectra were obtained using a JASCO-V570 spectrometer. Absorption spectra were measured with a TU-1901 UV-Vis spectrophotometer, and fluorescence measurements were carried out with the FP-6500 spectrofluorometer.
2.2 Crystallographic measurements and structure determination
A yellow single crystal of the title compound with dimensions of 0.30mm × 0.17mm × 0.15mm was mounted on a glass fiber in a random orientation.The data for the compounds were collected on a Bruker SMART Apex II CCD-based X-ray diffractometer equipped with a graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) using an ω-2θ scan mode in the range of 1.64≤θ≤25.02° (–11≤h≤11, –15≤k≤11, –46≤l≤46) at 298(2) K. The structure was solved by direct methods with SHELXS-97 and refined with SHELXL-97[18]. All the non-hydrogen atoms were refined by full-matrix least-squares techniques with anisotropic displacement parameters. The hydrogen atoms were geometrically fixed at the calculated positions attached to their parent atoms, and treated as riding atoms. The title compound crystallizes in monoclinic space group P21/n with a = 9.7685(8), b = 13.0519(13), c= 39.594(3) Å, β = 91.21(0)º, V = 5047.01(76) Å3, Z= 4, Mr= 1234.43, Dc= 1.625 g/cm3, F(000) = 1186,μ = 3.201 mm-1, S = 1.076, the final R = 0.0787 and wR = 0.1857 for 24216 observed reflections with I >2σ(I). And the selected bond lengths and bond angles as well as H bonds are given in Tables 1 and 2, respectively.
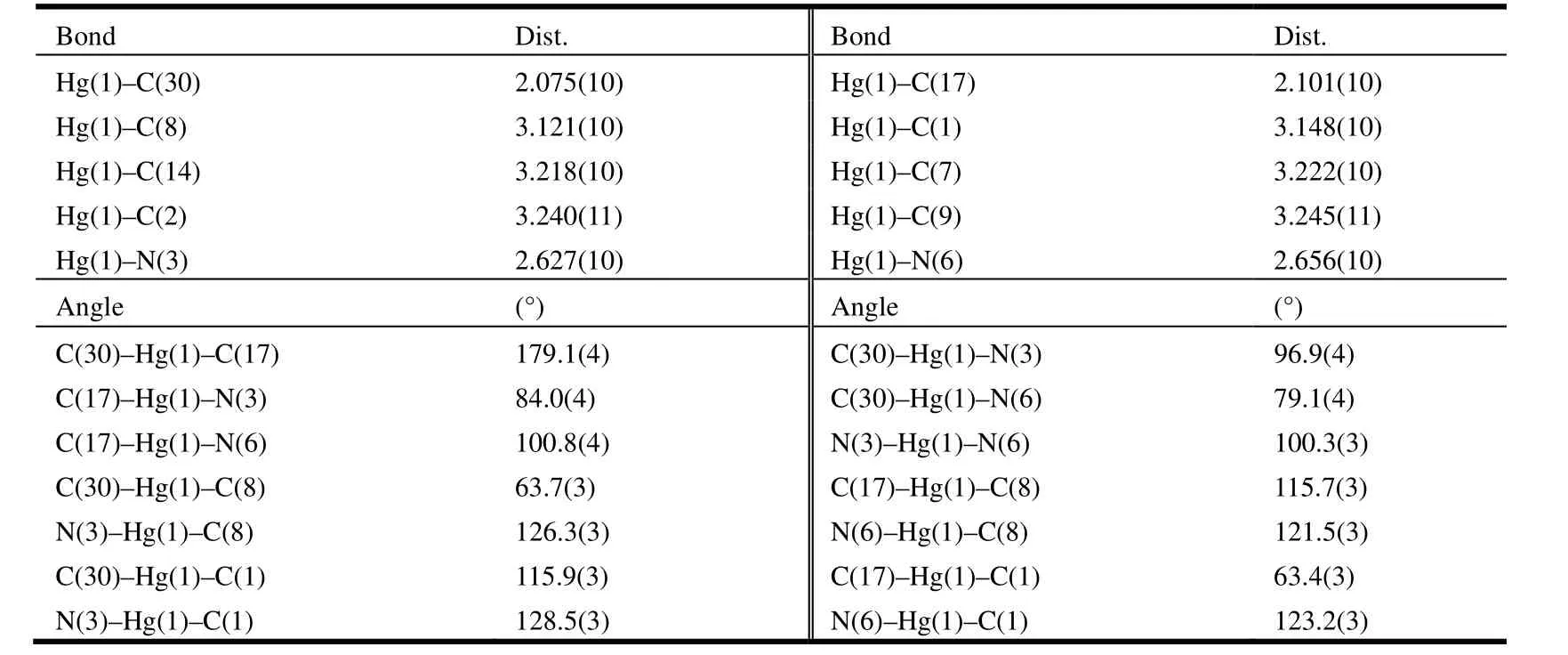
Table 1. Selected Bond Lengths (Å) and Bond Angles (º)
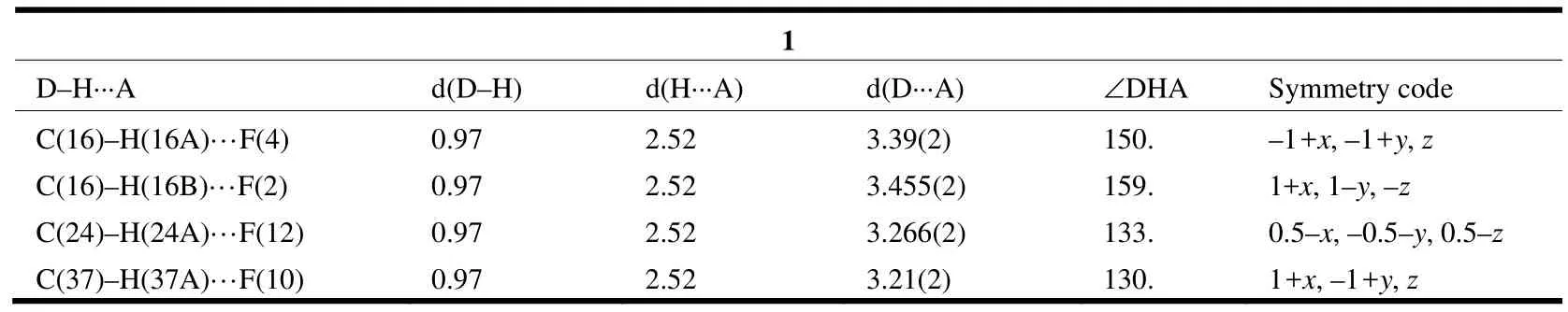
Table 2. Hydrogen Bond Lengths (Å) and Bond Angles (°) for 1
2.3 Synthesis of
N-(2-pyridylmethyl)benzimidazole N-(2-pyridylmethyl)benzimidazole was synthesized according to the procedure of literature[19]. The solution of benzimidazole (5.90 g, 50 mmol) was reacted with Na (1.15 g, 50 mmol) in THF (50 mL)until melt sodium disappears. 2-(Choromethyl) pyridine1 (6.35 g, 50 mmol) was added to the resulting solution at 0 ℃, and stirred for 24 h. Then, the white precipitate was removed by filtration through Celite.After removing the solvent in vacuo, the residue was washed with CH2Cl2. The combined organic phases were washed with H2O, dried with MgSO4, and concentrated in vacuo. The product crystallizes as white solid (Scheme 1). Yield: 8.20 g (78%). m.p.111.0~111.2 ℃.
2.4 Synthesis of 9,10-bis[3-(2-pyridylmethyl)-benzimidazolium-1-ylmethyl]anthracenebis(hexafluoro-phosphate)
To a suspension of N-(2-pyridylmethyl)benzi-midazole (2.09 g, 10.0 mmol) in toluene (100 mL)was added 9,10-bis(chloromethy1)anthracene (1.375 g, 5.0 mmol) in toluene (10 mL). After refluxing for 48 h, a white precipitate was formed and collected by filtration. Then, the solid was dissolved in methanol and an aqueous solution of ammonium hexafluorophosphate (1.63 g, 10.0 mmmol) was slowly added, after which the precipitate was collected by filtration and recrystallized from CH3CN and diethyl ether to give the title compound 2.80 g (yield 61.0%, Scheme 1). m.p. > 250 ℃.1H NMR (CD3COCD3, 600 MHz) δ: 9.42 (s, 2H, Hb),8.68~8.67 (dd, 4H, ArH), 8.45~8.43 (d, 2H, ArH),8.31 (d, 2H, ArH) 8.03~8.01 (d, 2H, ArH), 7.87~7.84 (m, 2H, ArH), 7.78~7.75 (m, 8H, ArH),7.53~7.51 (d, 2H, ArH), 7.28~7.26 (d, 2H, ArH),7.08 (s, 4H, Ha), 5.80 (s, 4H, Hc).13C NMR(CD3COCD3, 100 MHz) δ: 152.27, 149.45, 142.02,137.43, 132.10, 131.99, 128.11, 127.42, 125.63,124.43, 123.67, 122.46, 116.96, 114.15, 113.81,65.17, 51.04.
2.5 Synthesis of complex 1
A solution of ligand (0.365 g, 0.40 mmol) and Hg(OAc)2(0.128 g, 0.40 mmol) in CH3CN (20 mL)was refluxed under Ar atmosphere for 48 h. The solution was evaporated to dryness, and the residue was washed with water and diethyl ether. Recrystallization from CH3CN and diethyl ether produced a yellow solid, which was filtered and dried under vacuum. Yield: 0.41 g (92%). m.p. > 250 ℃. Elemental analysis for C48H41F12HgN9P2: Calcd. Anal.(%): C, 46.70; H, 3.35; N: 10.21. Found (%): C,46.62; H, 3.28; N, 10.09.1H NMR (CD3COCD3, 600 MHz) δ: 8.70~8.68 (dd, 4H, ArH), 8.56~8.55(d,2H, ArH), 8.31~8.30 (d, 2H, ArH), 8.00~7.98(m,2H, ArH), 7.93~7.90(m, 2H, ArH), 7.88~7.76(m,8H, ArH), 7.67~7.65(m, 2H, ArH), 7.34~7.33 (d,2H, ArH), 7.32 (s, 4H), 5.64(s, 4H).13C NMR(CD3COCD3, 100 MHz) δ: 154.6, 151.4, 149.4,146.4, 139.69, 134.29, 132.16, 128.60, 125.18,124.64, 116.92, 113.57, 54.9, 45.8.
3 RESULTS AND DISCUSSION
3.1 Synthesis and characterization
A convenient method for the formation of NHCs involves deprotonation of a benzimidazolium salt precursor. Reaction of 9,10-bis(chloromethyl)anthracene with N-(2-pyridylmethyl)-benzimidazole in THF at room temperature afforded the dibenzimidazolium salts in good yields. Treatment of the dibenzimidazolium chloro salts with NH4PF6in methanol affords the dibenzimidazolium hexafluorophosphate salts. Reaction of the ligand with Hg(OAc)2in a 1:1 molar ratio in CH3CN gave the title complex. Diffusion of diethyl ether into an acetonitrile solution of the title complex afforded yellow crystals of 1, which were stable in air and easily soluble in acetonitrile and acetone. The characteristic proton resonances observed in the1HNMR spectra of the ligand are the benzimidazolium N(CH)N proton (Hb) and two different kinds of methylene protons (Haand Hc) at 9.42, 7.08 and 5.80 ppm, respectively (Scheme 1). Upon coordination with mercury(II) ions, the carbenium proton (Hb)resonance disappeared and the chemical shifts of Hcmoved downfield by 0.24 ppm (from 7.08 to 7.32 ppm) as a result of the interaction of mercury atom and pyridine rings.
3.2 Structural description
A single-crystal X-ray diffraction study performed on 1 reveals the title complex crystallizes in monoclinic space group P21/n and the asymmetric unit contains one formula unit. As shown in Fig. 1, the whole molecule of 1 possesses a bridge shape. The mercury(II) center is bound to two carbene carbons from two benzimidazole groups and two nitrogen atoms from two pyridine groups. The geometry at the mercury atom coordinated with two 2-carbene carbons is nearly linear, with the C(30)–Hg(1)–C(17) bond angle of 179.1(4)º. The Hg–C bond lengths (Hg(1)–C(30) 2.075(1) Å, Hg(1)–C(17)2.101(1) Å) are shorter than the Hg–N(3) bond distances (Hg(1)–N(3) 2.627(1) Å, Hg(1)–N(6)2.656(1) Å). Such coordination mode leads to a remarkable structural change in the anthracene ring.For the four binding sites coordinated with mercury at the same time, the 9,10-carbon atoms of the anthracene ring are remarkably bent toward the metal core, out of the coplanar configuration, and form a dihedral angle of 11.1º between the side benzene rings in the anthracenyl units. The distances of the mercury ion to the six carbon atoms of the middle ring in anthracene are observed as follows:Hg(1)–C(8) = 3.121(1) Å, Hg(1)–C(1) = 3.148(1) Å,Hg(1)–C(14) = 3.218(1) Å, Hg(1)–C(7) = 3.222(1)Å, Hg(1)–C(2) = 3.240(1) Å and Hg(1)–C(9) =3.245(1) Å. The distance of the mercury atom to the centroid of anthracene is 3.101 Å, which is within the sum of van der Waals radii of carbon and mercury (3.25 Å). This distribution of Hg–C distances shows incipient η6coordination, which is comparable to that of the reported arene-metal complex[20]. The ligand, as a tetradentate coordinating unit, can easily clutch the mercury(II) ions, and that capacity should be credited to its two N-carbene functions to chelate the metal over the space above the anthracene π-face. Thus, a mercury-π interaction,which is enforced by the ligand structure, is observed. In the title compound 1, the intermolecular π···π interactions between anthracene rings (with offset face-to-face distance of 3.678(7) Å) direct two adjacent molecular moieties assembling into dimers,which forms 1D polymeric chains via intermolecular C(5)–H(5)···π (3.818(2) Å, 168(0)°) and C(41)–H(41)···π (3.589(2) Å, 146(0)°) inter- actions (Fig.2). Then, the 1D polymer could be further linked into a 3D supramolecular network structure via C–H···F interactions (Table 2) between the PF6-anions and methylene of the ligand (Fig. 3).

Scheme 1. Synthesis of the ligand and complex 1
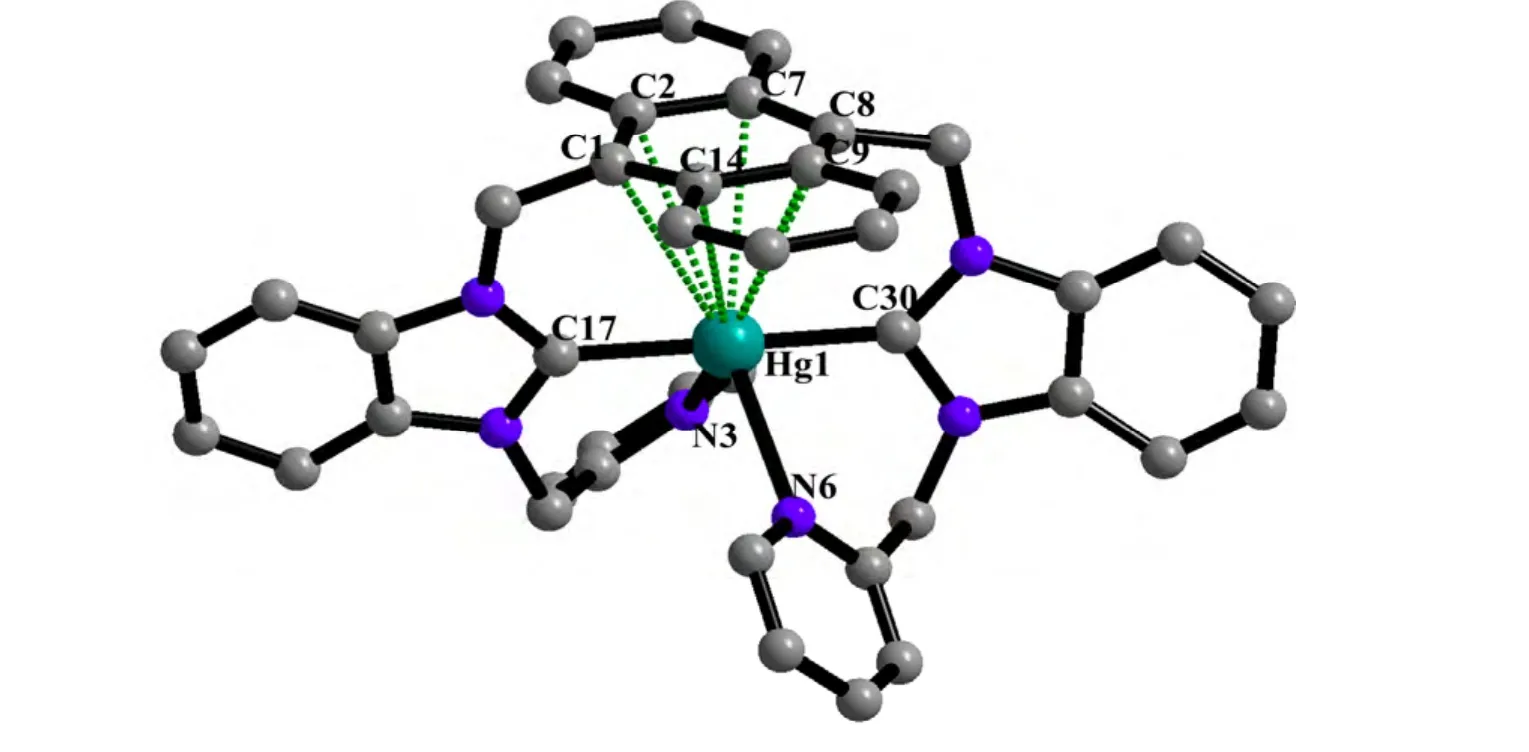
Fig. 1. View of the molecular structure of 1 (Hydrogen atoms have been omitted for clarity)
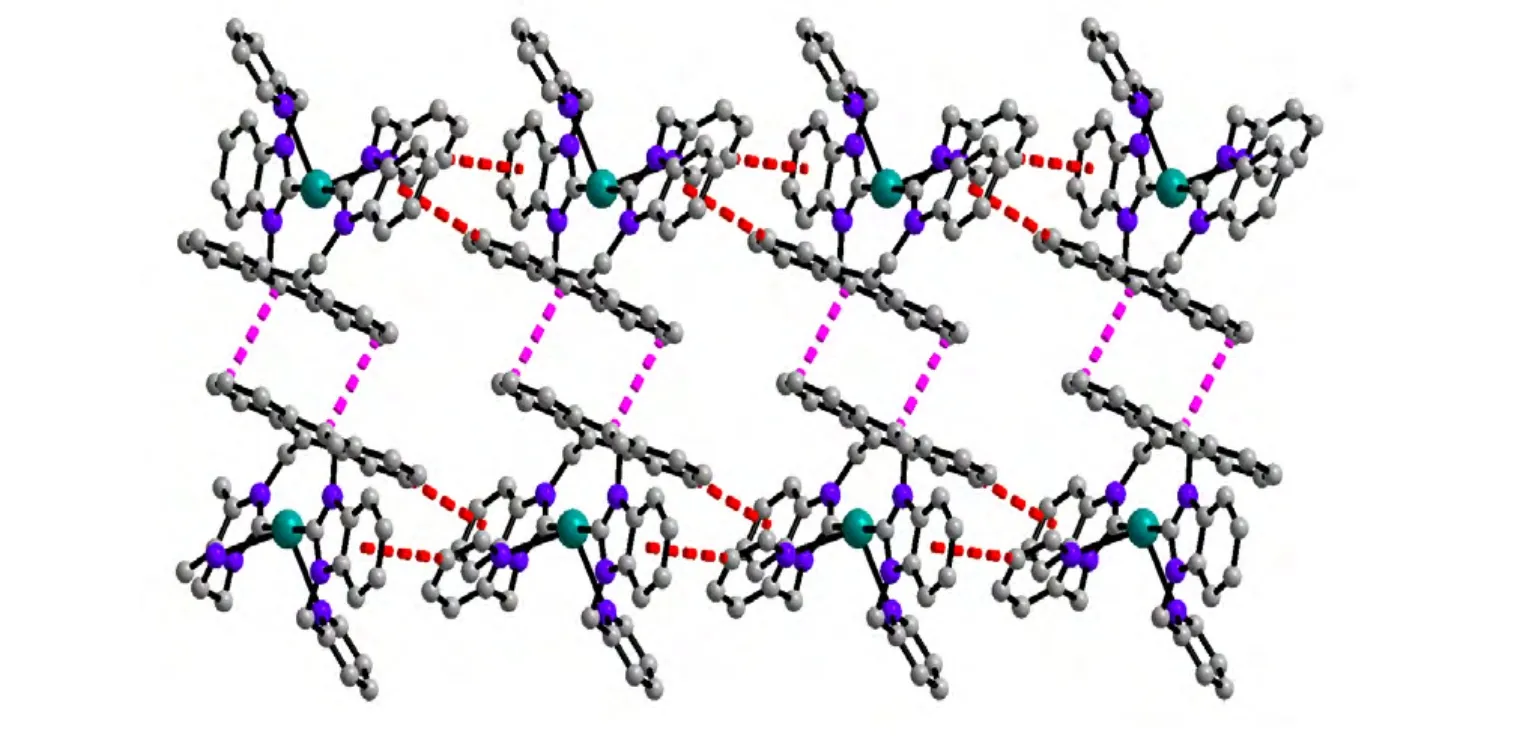
Fig. 2. View of the 1D polymeric chains constructed by intermolecular C–H···π and π···π interactions
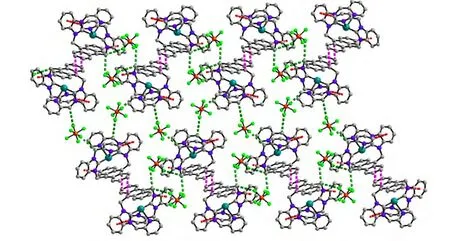
Fig. 3. View of the 3D supramolecular network along the a direction

Fig. 4. UV-vis absorption of the ligand and the title complex at 298 K in acetone (1.0 × 10-5 mol·L-1)

Fig. 5. Emission spectra of the ligand and the title complex at 298 K in acetone (1.0 × 10-5 mol·L-1)
3.3 Photoluminescence property
UV-vis and fluorescence spectroscopy had been employed to confirm the interaction of anthracene unit and mercury(II). The UV-vis (Fig. 4) of 1 shows that absorption peaks are similar to the UV-vis signals of free ligand, but with notable red shift of about 8 nm, corresponding to the bands centered at 339, 356, 375 and 396 nm, respectively, which are characteristic p-p* transitions of anthracene fluorophore. As indicated by recent work of fluorescence quenching based on the anthracene-tethered sensor[21,22],the red shift of absorption spectrum of 1 is probably a consequence of the shorter Hg-anthracene centroid distance, which could enhance the contribution of Hg to LUMO of 1 (charge transfer to Hg),thus increasing the energy gap between S0and S1.As shown in the fluorescence emission spectrum(Fig. 5), 1 exhibits a typical anthracene-based fluorescence emission in the range of 380~550 nm but much weaker than free ligand and exhibits slightly red shift of 4, 7 and 6 nm for the three main peaks from high energy to low energy region. The bathochromic shift in fluorescence spectrum of 1 should be caused by the same reason as Uv-vis spectrum[23]. The significant chelation enhanced quenching effect is mainly due to the large spinorbital coupling constant (ξ) of heavy metal ion Hg2+,and the short interaction between Hg and anthracene(π complex) will further facilitate the quenching effect[21,22].In addition, the fluorescent quantum yield (Φ) decreased from 0.91 to 0.16 and luminescent lifetime dropped from 1.19 ns to 0.31 ns, upon the free ligand coordinating to the mercury atom.
4 CONCLUSION
In summary, we have synthesized one bis(benzimidazolin-2-ylidene)arene-mercury(II) complex under a mild condition. Compared to the free ligand,the complex has exhibited a weaker photoluminescence in the visible region due to the ligand-tometal charge transfer process.
(1) Cook, T. R.; Zheng, Y. R.; Stang, P. J. Metal-organic frameworks and self-assembled supramolecular coordination complexes: comparing and contrasting the design, synthesis, and functionality of metal-organic materials. Chem. Rev. 2012, 113, 734–777.
(2) Steed, J. W.; Atwood, J. L. Supramolecular Chemistry, 2nd Ed.; Wiley, New York 2009.
(3) Steed, J. W.; Wallace, K. J. Advances in Supramolecular Chemistry Vol. 9 (Ed.: G. W. Gokel), Cerberus, New York 2003.
(4) Li, G.; Yu, W.; Ni, J.; Liu, T.; Liu, Y.; Sheng, E.; Cui, Y. Self-assembly of a homochiral nanoscale metallacycle from a metallosalen complex for enantioselective separation. Angew. Chem., Int. Ed. 2008, 47, 1245–1249.
(5) Li, G.; Yu, W.; Cui, Y. A homochiral nanotubular crystalline framework of metallomacrocycles for enantioselective recognition and separation. J.Am. Chem. Soc. 2008, 130, 4582–4583.
(6) Liu, T.; Liu, Y.; Xuan, W.; Cui, Y. Engineering homochiral metal-organic frameworks for heterogeneous asymmetric catalysis and enantioselective separation. Angew. Chem., Int. Ed. 2010, 122, 4215–4218.
(7) Xuan, W.; Zhang, M.; Liu, Y.; Chen, Z.; Cui, Y. A chiral quadruple-stranded helicate cage for enantioselective recognition and separation. J. Am.Chem. Soc. 2012, 134, 6904–6907.
(8) Murase, T.; Horiuchi, S.; Fujita, M. Naphthalene Diels-Alder in a self-assembled molecular flask. J. Am. Chem. Soc. 2010, 132, 2866–2867.
(9) Yuan, G.; Huo, Y.; Nie, X.; Jiang, H.; Liu, B.; Fang, X.; Zhao, F. Controllable supramolecular structures and luminescent properties of unique trimeric Zn(II) 8-hydroxyquinolinates tuned by functional substituents. Dalton Trans. 2013, 42, 2921–2929.
(10) Yuan, G.; Rong, L.; Qiao, X.; Ma, L.; Wei, X. Anion-controlled structures and luminescent properties of three Cd(II) complexes assembled by a 2-substituted 8-hydroxyquinoline ligand. CrystEngComm. 2013, 15, 7307–7314.
(11) Northrop, B. H.; Zheng, Y. R.; Chi, K. W.; Stang, P. J. Self-organization in coordination-driven self-assembly.Acc. Chem. Res. 2009, 42, 1554–1563.
(12) Fang, Y.; Murase, T.; Sato, S.; Fujita, M. Noncovalent tailoring of the binding pocket of self-assembled cages by remote bulky ancillary groups. J.Am. Chem. Soc. 2012, 135, 613–615.
(13) Li, S. S.; Northrop, B. H.; Yuan, Q. H.; Wan, L. J.; Stang, P. J. Surface confined metallosupramolecular architectures: formation and scanning tunneling microscopy characterization. Acc. Chem. Res. 2008, 42, 249–259.
(14) Chakrabarty, R.; Mukherjee, P. S.; Stang, P. J. Supramolecular coordination: self-assembly of finite two- and three-dimensional ensembles. Chem.Rev. 2011, 111, 6810–6918.
(15) Yao, L. Y.; Qin, L.; Xie, T. Z.; Li, Y. Z.; Yu, S. Y. Synthesis and anion sensing of water-soluble metallomacrocycles.Inorg. Chem. 2011, 50, 6055–6062.
(16) Lin, W. Q.; Leng, J. D.; Tong, M. L. A novel nanosized {Co16} metallamacrocycle incorporating four linear {Co4} subunits bridged by polytriazolate ligands. Chem. Commun. 2012, 48, 4477–4479.
(17) Du, J. L.; Hu, T. L.; Zhang, S. M.; Zeng, Y. F.; Bu, X. H. Tuning silver(I) coordination architectures by ligands design: from dinuclear, trinuclear, to 1D and 3D frameworks. CrystEngComm. 2008, 10, 1866–1874.
(18) Sheldrick G. M. SHELXTL V5.1 Software Reference Manual. Bruker AXS, Inc., Madison, Wisconsin, USA 1997.
(19) Sundberg, R. J.; Mente, D. C.; Yilmaz, I.; Gupta, G. Synthesis of bidentate and tridentate imidazole ligands.J. Heterocycl. Chem. 1977, 14, 1279–1281.
(20) Steckel, J. A. Ab initio modeling of neutral and cationic Hg-benzene complexes. Chem. Phys. Lett. 2005, 409, 4-6, 322–330.
(21) Lee, H.; Lee, H. S.; Reibenspies, J. H.; Hancock, R. D. Mechanism of “turn-on” fluorescent sensors for mercury(II) in solution and its implications for ligand design. Inorg. Chem. 2012, 51, 10904–10915.
(22) Lee, H.; Hancock, R. D.; Lee, H. S. Role of fluorophore-metal interaction in photoinduced electron transfer (PET) sensors: time-dependent density functional theory (TDDFT) study. J. Phys. Chem. A 2013. 117, 13345-13355.
(23) Yang, Y.; Wang, Y.; Peng, Y. A bifunctional, colorimetric and fluorescent probe for recognition of Cu2+and Hg2+and its application in molecular logic gate. Sci. China Chem. 2014, 57, 289–295.

- 结构化学的其它文章
- Synthesis and Crystal Structure of(Z)-2-Methyl-5,6-dihydrobenzo[d]thiazol-7(4H)-one O-Prop-2-yn-1-yl Oxime Derivatives①
- Synthesis and Crystal Structure of Cobalt(II) and Copper(II) Complexes Involving L-Aamino Alcohols
- Synthesis, Crystal Structure and Photoluminescence of a Three-coordinate Ag(I) Complex①
- Synthesis, Crystal Structure and Antimicrobial Activity of Ethyl 2-(1-cyclohexyl-4-phenyl-1H-1,2,3-triazol-5-yl)-2-oxoacetate①
- Synthesis, X-ray Crystallographic Analysis and Bioactivities of α-Aminophosphonates Featuring Pyrazole and Fluorine Moieties①
- Syntheses and Structural Characterizations of a Series of Capped Keggin Derivatives①