
吲哚-2-酮类衍生物的合成
2014-03-24 03:48康从民
化学与生物工程 2014年10期
宋 旸,康从民
(青岛科技大学化工学院,山东 青岛 266042)
吲哚-2-酮类衍生物是一种重要的杂环类化合物。很多抗癌药物分子中都含有吲哚-2-酮结构,如多靶点酪氨酸激酶抑制剂舒尼替尼(图1)[1-2]能够直接抑制血管内皮细胞的生长与增殖,从而发挥高效抗肿瘤作用[3-4]。因此,合成吲哚-2-酮类衍生物对研究新型抗癌药物具有重要的意义。

图1 舒尼替尼的结构式
吲哚-2-酮类衍生物的合成方法分为直接取代法和取代靛红还原法[5-7]。直接取代法条件苛刻,不易操作,而且产率较低;取代靛红还原法易操作,产率较高。文献报道的取代靛红合成方法较多[8-12]:如Martinet合成法[13],以芳香胺和丙酮二酸酯作用,然后水解、脱羧制得;Stoll合成法[14],以苯胺与草酰氯作用后,再经Lewis酸闭环制得;Gassman合成法[15],以苯胺与甲硫基乙酸酯作用,先闭环生成3-甲硫基-2-羟基吲哚,再经氧化制得;Sandmeyer合成法[16-17],以苯胺类衍生物与盐酸羟胺、水合氯醛反应生成异亚硝基乙酰苯胺类衍生物,然后在浓硫酸作用下发生分子内环合制得,原料易得、路线简短、条件温和、产率较高、产物较易分离,是较理想的取代靛红合成方法。
作者以苯胺类衍生物为起始原料,经Sandmeyer反应合成靛红类衍生物,再经水合肼还原,3位酮被还原成亚甲基,得到吲哚-2-酮类衍生物。合成路线如图2所示:

图2 吲哚-2-酮类衍生物的合成路线
1 实验
1.1 试剂与仪器
对甲苯胺、邻氯苯胺、盐酸羟胺,天津巴斯夫化工有限公司;对氯苯胺、80%水合肼,莱阳康德化工有限公司;对溴苯胺,国药上海试剂有限公司;水合氯醛,天津博迪化工有限公司;所用试剂均为分析纯。
Advance AV 500MHz型核磁共振仪(TMS为内标),瑞士Bruker公司;X4型数字显微熔点仪,巩义予华仪器有限公司。
1.2 方法
1.2.1异亚硝基乙酰邻氯苯胺的合成
在500 mL三口烧瓶中加入蒸馏水200 mL,置于油浴锅内,温度调至35 ℃,然后加入200 mL蒸馏水,开启机械搅拌;然后将12 g(72.5 mmol)水合氯醛、40 g(281 mmol)无水硫酸钠加入三口烧瓶中,待反应物完全溶解后,加入35 mL溶有6.38 g(50 mmol)对氯苯胺与6.0 mL浓盐酸的水溶液;用恒压滴液漏斗加入50 mL溶有9.76 g(148 mmol)盐酸羟胺的水溶液;滴加完毕后,将温度升至80 ℃下反应2 h,用薄层色谱检测(展开剂体积比:石油醚∶乙酸乙酯=1∶1);冷却至20 ℃,真空抽滤,水洗,得到淡黄色固体异亚硝基乙酰邻氯苯胺。
1.2.27-氯靛红的合成
在100 mL三口烧瓶中加入98%浓硫酸15 mL,置于油浴锅内,开启磁力搅拌,升温至50 ℃;搅拌下分批缓慢加入7.35 g(0.037 mol)异亚硝基乙酰邻氯苯胺,0.5 h加完,控制温度在50~60 ℃;加毕,升温至70 ℃反应1 h;用薄层色谱鉴定无原料点后(展开剂体积比:石油醚∶乙酸乙酯=2∶1),停止反应,冷却至室温,搅拌下缓慢倒入50 g碎冰中,磁力搅拌下水解0.5 h,然后冰浴静置0.5 h;抽滤,滤饼用水洗涤后溶于160 mL 10%NaOH溶液中,用稀盐酸调pH=3(析出沉淀);抽滤(除去黑色杂质),滤液用稀盐酸调pH=2(有大量固体析出);抽滤,滤饼用水洗涤,干燥得桔红色产物7-氯靛红11.72 g。
1.2.37-氯吲哚-2-酮的合成
将20.6 mL 80%水合肼加入250 mL三口烧瓶中,搅拌下加入7-氯靛红2.2 g,升温至100 ℃;补加7-氯靛红6.6 g,升温至110 ℃反应4 h;再升温到120 ℃,将反应液抽滤,收集固体,将固体悬浮于20 mL水中,搅拌下用浓盐酸调pH<3,于室温下搅拌12 h;抽滤,用大量水洗涤至滤饼基本无色,烘干,得到棕色固体7-氯吲哚-2-酮。
同法合成5-氯吲哚-2-酮、5-甲基吲哚-2-酮和5-溴吲哚-2-酮。
2 结果与讨论
2.1 中间体及目标化合物的表征
(1)异亚硝基乙酰邻氯苯胺:产率93.1%,熔点172 ℃(文献[18]值172~173 ℃)。
7-氯靛红:产率71%,1HNMR (DMSO),δ:11.47(s,1H,N-H),7.65(d,1H,Ar-H),7.48(d,1H,Ar-H),7.09(t,1H,Ar-H)。
7-氯吲哚-2-酮:产率52.1%,1HNMR (DMSO),δ:10.80(s,1H,N-H),7.23(d,1H,Ar-H),7.18(d,1H,Ar-H),6.97(t,1H,Ar-H),2.08(s,2H,Ar-CH2)。
(2)异亚硝基乙酰对氯苯胺:产率94%,熔点175 ℃(文献[18]值174~175 ℃)。
5-氯靛红:产率77%,1HNMR (DMSO),δ:12.24(s,1H,N-H),7.73(d,1H,Ar-H),7.64(s,1H,Ar-H),7.39(d,1H,Ar-H)。
5-氯吲哚-2-酮:产率49.8%,熔点195 ℃(文献[18]值195~197 ℃)。
(3)异亚硝基乙酰对甲苯胺:产率86.2%,1HNMR (DMSO),δ:12.15(s,1H,N-H),10.10(s,1H,Ar-H),7.65(s,1H,CH=N),7.57(d,2H,Ar-H),7.13(d,2H,Ar-H),2.25(s,3H,Ar-CH3)。
5-甲基靛红:产率79.3%,熔点186 ℃(文献[18]值185~187 ℃)。
5-甲基吲哚-2-酮:产率59.3%,熔点174 ℃(文献[18]值173~174 ℃)。
(4)异亚硝基乙酰对溴苯胺:产率90.9%,熔点195 ℃(文献[18]值195~197 ℃)。
5-溴靛红:产率81%,1HNMR (DMSO),δ:10.49(s,1H,N-H),7.24(s,1H,Ar-H),7.20(d,1H,Ar-H),6.80(d,1H,Ar-H)。
5-溴吲哚-2-酮:产率65.2%,熔点220 ℃(文献[18]值219~221 ℃)。
2.2 取代基团对反应时间和产率的影响
由于苯环上取代基团的不同,肟的合成难易程度也不同。在合成异亚硝基乙酰苯胺类衍生物的过程中,当苯环上取代基团为吸电子基团(如对氯苯胺和对溴苯胺)时,由于吸电子效应,使得胺上孤对电子密度减小,反应较难进行,反应时间较长,温度较高,优点是副产物较少,产率较高;当苯环上取代基团为供电子基团(如甲基)时,反应较容易,反应时间较短,温度较低,缺点是副产物较多,且多为黄色油状液体,冷却后为黄色固体,与产物混在一起,不易除去。
综合考虑,本实验采用适当降低温度的方法,尽量减少副反应的发生。对于生成的副产物,可以用水作为重结晶溶剂,并加入少量活性炭作为吸附剂进行纯化。该方法较文献[8-9]报道的正己烷+丙酮混合重结晶更经济环保。
2.3 还原剂的选择
靛红类衍生物还原制备吲哚-2-酮类衍生物的方法主要有克莱门森还原法和黄鸣龙还原法。由于靛红类衍生物存在酰胺键,当遇到盐酸等强酸时会水解,导致产率降低甚至实验失败;但也不能在强碱性环境下反应,否则会导致酰胺键水解从而影响产率。
综合考虑,本实验采用加入弱碱水合肼的方法来进行还原,一方面,水合肼可以作为还原剂参与反应,另一方面可提供适当的弱碱性环境以促进反应的进行。还原反应分两步进行:
第一步:靛红3位的羰基和肼反应,形成腙,该步反应较容易,反应迅速。
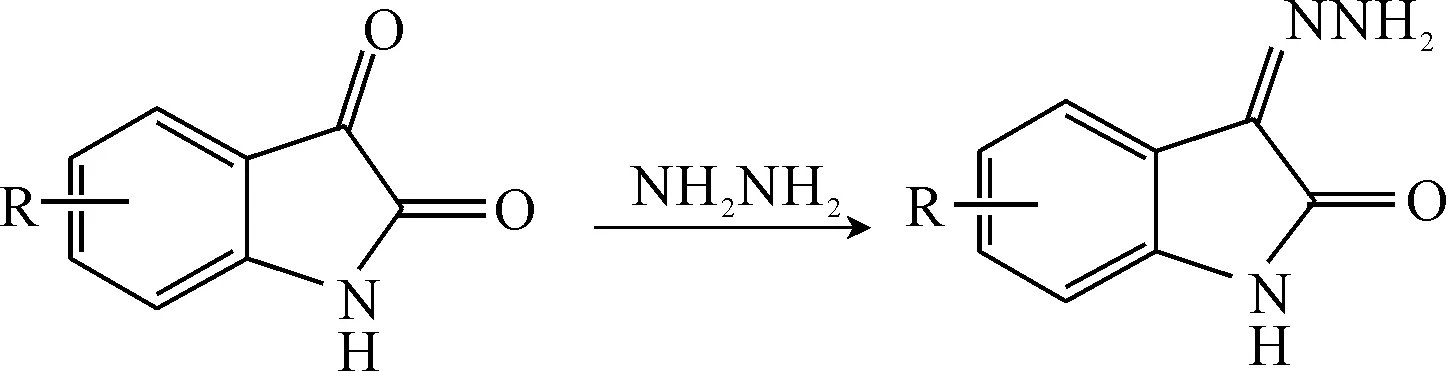
第二步:在较高温度下,腙转换为亚甲基。

2.4 7-氯吲哚-2-酮合成过程中的特殊性
(1)在合成7-氯靛红的过程中,加入异亚硝基乙酰邻氯苯胺的速度一定要慢,少量多次加入,同时控制温度在50~60 ℃。这是因为,加入速度过快会导致硫酸升温过快,容易产生副反应,而且生成黄色烟雾喷出,不利于安全。反应时间过长容易产生副反应,生成不易去除的黑色絮状聚合物,降低产率;反应时间过短,反应进行的不完全,产率也会降低。因此,本实验选择1 h作为较适宜的反应时间。靛红类衍生物粗产品采用NaOH溶解、HCl析出的方法进行纯化[8]。
(2)水合肼还原7-氯靛红生成7-氯吲哚-2-酮的过程中,要先加入少量的7-氯靛红,然后升温到100 ℃后再加入剩余的7-氯靛红。如果一次性加入,易生成淡黄色泡沫状物质并溢出,这是因为,7-氯靛红的3位羰基与过量的肼反应生成腙,腙分解生成的氮气使得产物呈泡沫状而溢出。
3 结论
以苯胺类衍生物为起始原料,先经Sandmeyer合成得到靛红类衍生物,再经水合肼还原得到系列吲哚-2-酮类衍生物:7-氯吲哚-2-酮、5-氯吲哚-2-酮、5-甲基吲哚-2-酮和5-溴吲哚-2-酮,产率分别为52.1%、49.8%、59.3%和65.2%。
参考文献:
[1]MOTZER R J,MICHAELSON M D,REDMAN B G,et al.Activity of SU11248,a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor,in patients with metastatic renal cell carcinoma[J].J Clin Oncol,2006,24(1):16-24.
[2]FAIVER S,DEMETRI G,SARGENT W,et al.Molecular basis for sunitinib efficacy and future clinical development[J].Nat Rev Drug Discov,2007,6(9):734-745.
[3]方正,杨照,鲍书馨,等.舒尼替尼的合成工艺研究[J].化学试剂,2010,32(1):82-84.
[4]ROBERTS L R.Sorafenib in liver cancer—just the beginning[J].N Engl J Med,2008,359(4):420-422.
[5]韩爽,李东风,吴丛梅,等.吲哚喹唑啉衍生物的合成与表征[J].化学试剂,2011,33(10):883-886.
[6]沈学全,吴永龙,钱海均.5-氟-吲哚-2-酮的合成工艺研究[J].化工时刊,2012,26(4):29-31.
[7]闵真立,姜凤超,张奇.3-取代吲哚酮类化合物的合成及抗肿瘤活性[J].中国药物化学杂志,2005,15(3):129-132.
[8]廖玉华,陈文兵,张晓梅,等.靛红衍生物的合成[J].合成化学,2009,17(5):612-614,636.
[9]张晓飞,刘华业,高文涛.取代靛红改进方法的合成[J].渤海大学学报(自然科学版),2009,30(3):212-216.
[10]颜耘,赵萍,孙广龙,等.5-硝基靛红衍生物的合成[J].精细化工中间体,2011,41(5):32-34.
[11]张婷.卤素取代靛红衍生物的合成方法[J].河北化工,2011,34(7):15-17.
[12]俞鸣烽,张爱英,刘增路,等.靛红衍生物的制备[J].中国医药工业杂志,2008,39(5):332-333.
[13]WELSTEAD W J,MORAN H W,STAUFFER H F,et al.Antiinflammatory agents.1.Synthesis and antiinflammatory activity of 2-amino-3-benzoylphenylacetic acid[J].J Med Chem,1979,22(9):1074-1079.
[14]CHRISTIAN R,EMILE B.Ethyl(4-N-acylaminopyridin-3-yl)glyoxylate and 5-azaisatin as new synthons for a route to various new polyheterocycles[J].Heterocycl Chem,1997,34(2):441-444.
[15]GASSMAN P G,CUE J.A general method for the synthesis of isatins[J].J Org Chem,1977,(42):1344-1348.
[16]RICE K C,BOONE B J,RUBIN A B.Synthesis,antimalarial activity,and phototoxicity of some benzo(h) quinoline-4-methanols[J].J Med Chem,1976,19(7):887-892.
[17]张陈,陆涛.2-取代-4(3H)-喹唑啉酮类化合物合成研究进展[J].精细化工中间体,2006,36(5):12-15.
[18]李占成,金云舟,高博.5-取代吲哚-2-酮的合成[J].合成化学,2012,20(1):119-122.
猜你喜欢
氯碱工业(2022年6期)2022-11-21
昆明医科大学学报(2021年8期)2021-08-13
化工环保(2021年2期)2021-04-25
云南化工(2020年5期)2020-06-12
盐科学与化工(2019年11期)2019-12-04
天然产物研究与开发(2018年7期)2018-08-21
天然产物研究与开发(2018年4期)2018-05-07
橡胶科技(2016年6期)2016-02-24
中国塑料(2015年10期)2015-10-14
天然产物研究与开发(2014年5期)2014-04-27
