
Examination of Huntington’s disease in a Chinese family
2014-03-24 03:25:47MingxiaYuXiaogaiLiSanyunWuJiShenJianchengTu
中国神经再生研究(英文版) 2014年4期
Mingxia Yu, Xiaogai Li, Sanyun Wu, Ji Shen, Jiancheng Tu
Examination of Huntington’s disease in a Chinese family
Mingxia Yu, Xiaogai Li, Sanyun Wu, Ji Shen, Jiancheng Tu
Mingxia Yu and Xiaogai Li contributed equally to this work.
We report brain imaging and genetic diagnosis in a family from Wuhan, China, with a history of Huntington’s disease. Among 17 family members across three generations, four patients (II2, II6, III5, and III9) show typical Huntington’s disease, involuntary dance-like movements. Magnetic resonance imaging found lateral ventricular atrophy in three members (II2, II6, and III5). Moreover, genetic analysis identi fi ed abnormally ampli fi ed CAG sequence repeats (> 40) in two members (III5 and III9). Among borderline cases, with clinical symptoms and brain imaging features of Huntington’s disease, two cases were identi fi ed (II2 and II6), but shown by mutation analysis for CAG expansions in the important transcript 15 gene, to be non-Huntington’s disease. Our fi ndings suggest that clinical diagnosis of Huntington’s disease requires a combination of clinical symptoms, radiological changes, and genetic diagnosis.
nerve regeneration; neurodegenerative disease; Huntington’s disease; clinical symptoms; imaging; genetic diagnosis; IT15 gene; CAG repeat; neural regeneration
Funding: This study was supported by the Fundamental Research Funds for the Central Universities, No. 20100141110017, 20103030201000217 and 201130302020008.
Yu MX, Li XG, Wu SY, Shen J, Tu JC. Examination of Huntington’s disease in a Chinese family. Neural Regen Res. 2014;9(4):440-446.
Department of Clinical Laboratory Medicine & Center for Gene Diagnosis, Zhongnan Hospital of Wuhan University, Wuhan, Hubei Province, China
Introduction
Huntington’s disease is an autosomal-dominant inherited neurodegenerative disease with a distinct phenotype[1-3]. Clinical symptoms include progressive abnormal dancelike movements, mental dysfunction, and dementia. Brain imaging shows mainly caudate nucleus pathology and cortical atrophy[4-5]. Huntington’s disease prevalence worldwide, ranges from 5 to 10 per 100,000 people (0.05–0.1%) with signi fi cant morbidity in Western Europe and North America[6-7]. Typically, Huntington’s disease occurs during the third to fifth decade of life, showing slow progression and with affected individuals generally dying after 15 to 20 years. An earlier age of onset induces more rapid disease progression. Prevalence is approximately equal for men and women[8-10]. The clinical and hereditary aspects of Huntington’s disease were fi rst described by George Huntington in 1872, although the disease was known before this. In the 1980s and 1990s, development and application of exon amplification and cDNA cloning technology led to identi fi cation of important transcript 15 (IT15), the gene responsible for Huntington’s disease, by a Huntington’s chorea collaborative research team[11-13]. For suspected Huntington’s disease patients, gene screening, neuropathological detection, radiological change, and laboratory tests, coupled with complete family histories, effectively improved the diagnosis rate[14-16]. Predictive testing is available for individuals who are at risk of carrying the Huntington’s disease gene. Previous studies show that 3% of individuals with Huntington’s disease occur without a family history. Although patients with a family history have more typical clinical symptoms, signs, and pathological changes, as well as an unambiguous clinical diagnosis, other diseases with dance-like movements, e.g., dentatorubral-pallidoluysian atrophy, spinocerebellar ataxia type 17, Huntington’s disease-like-2, and neuroferritinopathy, are dif fi cult to identify and distinguish from Huntington’s disease[16-21]. To improve detection accuracy and speci fi city, combined analysis of clinical symptoms, imaging changes, and genetic tests are needed. If typical clinical Huntington’s disease symptoms and imaging changes are absent, it is particularly important to make an accurate gene diagnosis for excluding non-Huntington’s disease neurodegenerative diseases. A reasonable strategy is to fi rst test if the Huntington’s disease mutation is present.
In the past two decades, accumulating evidence from collaborative scientific research has found that instable CAG expansion within gene sequences is the genetic basis for Huntington’s disease development. Genetic dosage (CAG repeat length) of IT15 is closely linked with clinical features (e.g., age of onset). The mutant protein encoded by the pathogenic gene has also been identified. These findings provide insight into the mechanisms underlying neuronal degeneration in Huntington’s disease, and enabled development of genetic animal models[22-25]. Predictive testing for Huntington’s disease in individuals with inherited mutantgenes and abnormally expanded CAG repeats, provides the opportunity to develop preventive Huntington’s disease therapies, potentially delaying or even inhibiting occurrence and development of Huntington’s disease[26-27].
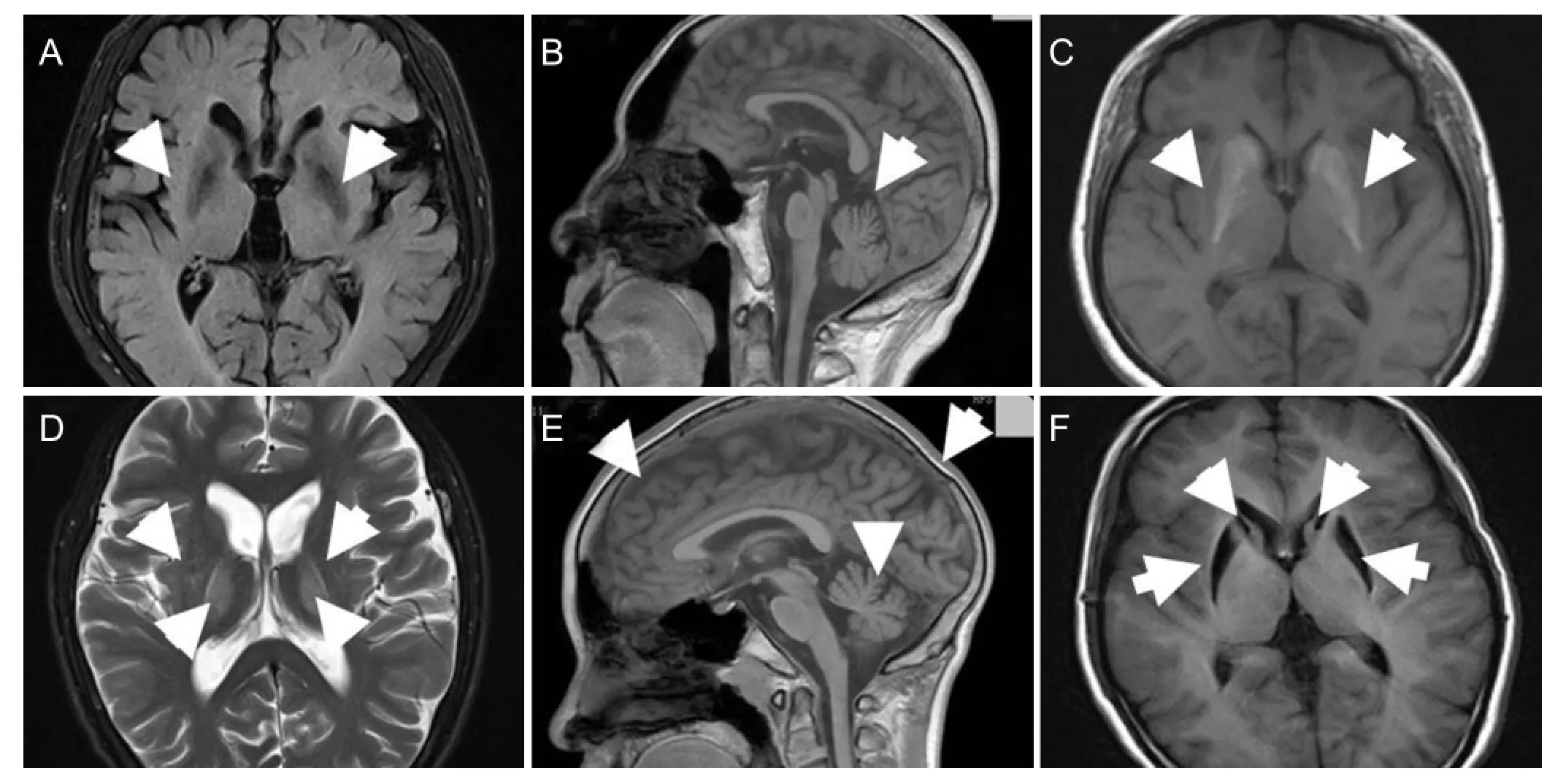
Figure 1 Brain MRI examination of four Huntington’s disease cases from the identi fi ed Huntington’s disease family.
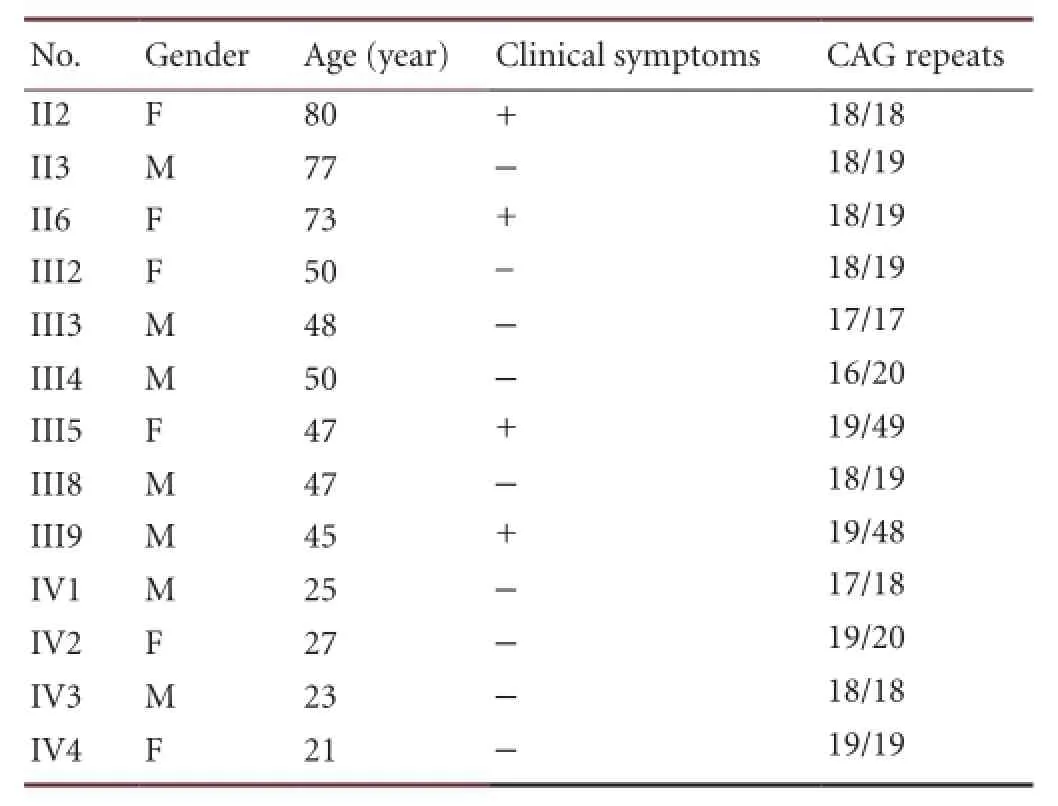
Table 1 Gene analysis of the Huntington’s disease family from Wuhan, Hubei Province, China
In this study, we investigated a Chinese family from Wuhan, Hubei Province, with Huntington’s disease. By mutation screening for CAG repeats in the Huntington’s disease-associated candidate gene, IT15, using reverse transcription-polymerase chain reaction, T-A cloning, and sequencing, we provide a global analysis incorporating clinical symptoms, imaging examinations, and gene diagnosis, of Huntington’s disease within this family.
Results
Quantitative analysis of participants
A four-generation family comprising 22 individuals all from the Han population, was identi fi ed. Thirteen members were recruited, with fi ve deaths and four uncooperative cases. Informed consent was obtained from all participating family members or their parents, after explaining the nature and possible consequence of the study. Detailed information of the subjects is shown (Table 1).
Abnormal radiological changes in II2, II6, and III5
Brain MRI images for four family members are shown (Figure 1). In the proband (III5), axial weighted images showed bilateral cerebral cortex and caudate nucleus atrophy, and abnormal lenticular nucleus signals. Cerebellar atrophy was observed in sagittal images. For III9, abnormal signals appeared only in the left lateral caudate nucleus head, and there was no signi fi cant ventricular expansion or sulci thickening. Lateral ventricular expansion was observed in II2 and II6. In II2, axial images showed widened sulci and ventricular enlargement, both brain atrophy indicators. Sagittal images revealed cerebellar atrophy. In II6, lateral ventricular expansion was observed, indicating caudate nucleus head atrophy. Abnormal radiological changes were found in II2, II6,and III5. No obvious brain parenchyma abnormalities were observed in III9.
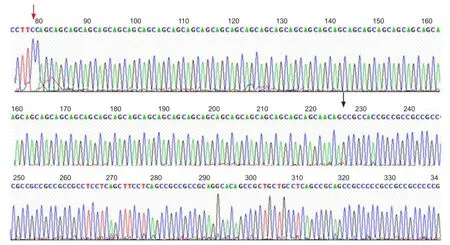
Figure 3 Important transcript 15 (IT15) gene sequencing results in the proband.

Figure 2 Important transcript 15 (IT15) gene expression determined by reverse transcription-polymerase chain reaction in the identi fi ed Huntington’s disease family.
III5 and III9 areIT15gene carriers
Reverse transcription-polymerase chain reaction showed abnormal 400 bp bands present in III5 and III9. Surprisingly, in II2 and II6 who presented with Huntington’s disease features, only a normal band of 150–200 bp was detected, which was also present in the other individuals (Figure 2). Overall, our results show that III5 and III9 are IT15 mutant gene carriers, yet there is no evidence indicating II2 and II6 are Huntington’s disease patients.
Forty-nine CAG repeats identi fi ed in the proband
Sequencing analysis identified 49 CAG repeats in the proband (Figure 3). CAG repeats for every participant are summarized (Table 1). III5 and III9 have one Huntington’s disease allele with > 40 CAG repeats, and one normal allele. This shows consistency between our DNA sequencing results and polyacrylamide gel electrophoresis analysis.
Discussion
Huntington’s disease is an autosomal dominant disorder characterized by progressive loss of medium spiny neurons in the striatum and, to a lesser extent, pyramidal neurons in the cortex[28-30]. The Huntington’s disease gene, IT15, was identi fi ed in 1993 and is located on chromosome 4p16.3[31-32]. The disease is caused by abnormal CAG sequence expansion within IT15, which is related to the age of disease onset[33-34]. The average age of Huntington’s disease onset is between 20 and 50 years, but adults of 35–40 years can be affected[35-38]. Klempíř et al.[39-40]found no evidence for the normal allele playing a role in modifying the age of onset.
A normally sized IT15 allele has a CAG repeat region of ≤26 repeats, generating a non-disease phenotype. Individuals with CAG repeats in the intermediate (27–35) range are considered at risk for Huntington’s disease development[41-44]. Individuals with 36–39 CAG repeats may develop Huntington’s disease during their lifetime, and their children are at greater risk of developing the disease. There are no reports of Huntington’s disease absence in any individual with > 40 CAG repeats[45-48].
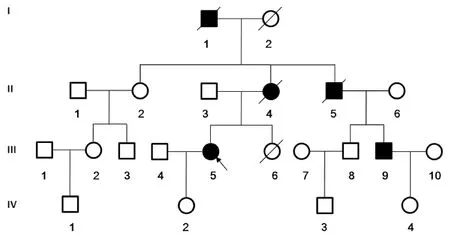
Figure 4 Pedigree of the Huntington’s disease family.
The proband in our study suffered from confusion, unresponsiveness, and memory loss. The worsening symptoms suggested progressive cerebral cortex atrophy. Genetic test results showed the CAG repeat sequence (at the 5′ end of the IT15 gene coding region) was 49, above the normal range, indicating she suffered from Huntington’s disease. The mother and uncle of the proband both had involuntary head and neck twitching and appeared physically uncoordinated. The cousin displayed clumsiness and was diagnosed with tetany at 35 years of age, schizophrenia at 38 years, with a high index for suspicion of Huntington’s disease.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis shows only one band in healthy controls, with subjects suspected of Huntington’s disease having an extra band between 350–400 bp. According to the gene sequence, the CAG repeat number = (PCR product length–164)/3, with an error of ± 1–2 repeats[49-51]. The CAG repeat number in the proband was 62–78. Agarose gel electrophoresis detection of the CAG repeat number is affected by CCG repeats in the primers, designed for CAG repeats and adjacent CCG repeats, which also exist as polymorphisms. Therefore, we used DNA sequencing to accurately determine the number of CAG repeats. In this family, III5 and III9 have 49 and 48 CAG repeats, respectively, in IT15, and both are heterozygous, carrying a Huntington’s disease and normal allele (the CAG repeat number was 16–20 in the normal allele). Gel electrophoresis test results can only be used for a preliminary decision. The fi nal genetic diagnosis is dependent on gene sequencing.
In this study, mutational analysis of the IT15 gene indicated abnormal extension with repeated CAG number, thereby indicating Huntington’s disease. However, among family members, there is signi fi cant difference in clinical manifestations, brain imaging pathology, and presence or absence of the IT15 gene mutation. III5 and III9 have > 40 CAG repeats, yet their clinical features are distinct. III9 does not show any obvious caudate nucleus atrophy or paracele enlargement. Brain scans show ataxic FLAIR signal in only the caudate nucleus head from the right paracele hook angle, but with no obvious ventricular expansion. II2 and II6 both show involuntary choreic movements and paracele expansion by brain CT and MRI scans, but no IT15 gene mutation on genetic diagnosis. Repeat rates of the CAG number in these two individuals are 18 and 19, and Huntington’s disease can be ruled out. A diagnosis of Huntington’s disease-like-2 could be made from detection of additional genes, including DRPLA, JPH3, and TBP, whose mutation results in the neural system movement disorders, dentatorubral-pallidoluysian atrophy, Huntington’s disease-like-2, and spinocerebellar ataxia type 17, respectively[52-58]. No IT15 mutations are found in 1% of patients suffering from clinical features of Huntington’s disease[36,59-60].
In summary, Huntington’s disease diagnosis should not be determined only from clinical features and brain imaging alterations, but also by genetic diagnosis[61]. Recommended genetic tests should include IT15, DRPLA, JPH3, and TBP[62], especially for patients whose main clinical feature is progressively more severe choreic movements, rather than obvious dementia and mental anomalies.
Subjects and Methods
Design
A familial, clinical case report.
Time and setting
Experiments were performed at the Department of Clinical Laboratory Medicine & Center for Gene Diagnosis, Zhongnan Hospital of Wuhan University, Wuhan, China from July 2011 to October 2012.
Subjects
A four-generation family was recruited from Zhongnan Hospital (Wuhan, Hubei Province, China). From the 22 family members, there were 13 participants (Figure 4). Detailed medical histories were obtained by interviewing all family members.
Three members are dead (I1, II4, and III5). I1 is the proband’s grandfather, with disease onset at 42 years old and dead by 45. II4, the proband’s mother, had disease onset at 45 years old and died at 60. II5, the proband’s maternal uncle, had disease onset at 36 years old and was dead by 40. Before death, all individuals had severe involuntary movements and were bedridden after losing the ability to walk independently.
Among the other participants, the symptoms of II2, II6, and III9 are very similar to the proband (III5). The proband’s cousin, III9, became symptomatic at 35 years old, and has mild involuntary movements in the fingers and wrists, which gradually spread to the face and shoulders. Increasing symptom severity led to grimacing, involuntary shrugging and dancing of the upper limbs, and an irritable personality. No other medical and surgical abnormalities were found by neurological examination.
Experiments were performed in strict accordance with the Declaration of Helsinki tenets. Written informed consent was obtained from all participating adult individuals and parents of children under 16 years old.
Methods
Clinical evaluation
The proband (III5), a 47-year-old married female with one daughter, presented with increasing irritability and spontaneous involuntary limb action in the past 2 months, although the symptoms started 2 years ago. Clinical manifestations include involuntary choreic movements of the upper limbs, an unsteady gait, lisp, and cognitive decline. Neurological examination revealed unconsciousness, involuntary movements, and muscular atrophy of the four extremities, slightly lower muscle tension, muscle strength grade 4, ankle-knee-tibia test (+), Romberg sign (+), and no pathological re fl ex. Urine and blood tests, liver and kidney function, electrolyte levels and electrocardiograms were normal. MRI of the head revealed severe brain atrophy. The other family members had similar medical histories.
Brain MRI analysis
Images were obtained using the 1.5 T MRI system (Philips Corporation, Amsterdam, Holland), with transverse T1- and T2-weighted scans.
Genetic analysis
Peripheral venous blood samples were collected from all study subjects. Genomic DNA was extracted from peripheral blood leukocytes using standard procedures. The IT15 gene coding sequence (GenBank NG_009378) was PCR amplified. Coding sequences were: IT15 forward, 5′-CCC AGA GCC CCA TTC ATT GCC-3′; IT15 reverse, 5′-GGC GGC GGT GGC GGC TGT TGC-3′ (Invitrogen, Shanghai, China). Amplifications were performed in 25 µL reactions containing 200 ng of genomic DNA with 2 × GC buffer, 10 pmol primers, 10 mmol/L dNTP, and 1 U Takara LA Taq polymerase (Takara Bio Ltd., Otsu, Shiga, Japan). PCR conditions were: initial denaturation at 94°C for 5 minutes, followed by 30 cycles of denaturation at 94°C for 30 seconds, annealing at 60°C for 45 seconds and extension at 72°C for 1 minute, with a fi nal extension for 10 minutes. PCR products were resolved by 8% polyacrylamide gel electrophoresis, and following Evans blue staining, images were obtained using the DP-JS-680B Automatic Gel Imaging Device (Shanghai Peiqing, China).
PCR products from the proband and one unaffected family member were puri fi ed from agarose gels using a gel extraction kit, ligated into the pMD18-T vector (Takara Bio Ltd.), and sequenced using the ABI Genetic Analyzer 3730 (Invitrogen). Sequencing results were analyzed using Chromas 2.3 software (Technelysium Pty Ltd., South Brisbane, Australia) and compared against the NCBI database reference sequence (http://www.ncbi.nlm.nih.gov/nuccore/ NM_002111.6).
Author contributions:Yu MX designed and revised the article. Li XG conducted the experiment, performed statistical analysis and wrote the article. Wu SY and Shen J collected clinical data. Tu JC was responsible for funds, supported and guided the study. All authors approved the final version of the manuscript.
Con fl icts of interest:None declared.
Peer review:The article discussed the characteristics of Huntington’s disease from familial inheritance, clinical features, imaging changes, and genetic analysis. These clinical Huntington’s disease cases especially with a complete family were difficult to collect, also the experimental data was relatively intact, the description for clinical symptom was clear, and genetic analysis results were reliable, showing that on the diagnosis of Huntington’s chorea, it is very important to use a variety of diagnostic methods.
[1] Phillips W, Shannon KM, Barker RA. The current clinical management of Huntington’s disease. Mov Disord. 2008;23(11):1491-1504.
[2] Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci. 2008;31(10):521-528.
[3] Ha AD, Fung VS. Huntington’s disease. Curr Opin Neurol. 2012; 25(4):491-498.
[4] Rosas HD, Salat DH, Lee SY, et al. Cerebral cortex and the clinical expression of Huntington’s disease: complexity and heterogeneity. Brain. 2008;131(Pt 4):1057-1068.
[5] Beste C, Stock AK, Ness V, et al. A novel cognitive-neurophysiological state biomarker in premanifest Huntington’s disease validated on longitudinal data. Sci Rep. 2013;3:1797.
[6] Gonzalez-Alegre P, Afifi AK. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol. 2006;21(3):223-229.
[7] Pogledić I, Relja M. Huntington’s disease. Lijec Vjesn. 2012;134(11-12):346-350.
[8] Politis M, Pavese N, Tai YF, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp. 2011; 32(2):258-270.
[9] Fernández Hawrylak M, Grau C, Trigo P. The impact of Hungtinton’s disease on the family. An Sist Sanit Navar. 2012;35(2):295-307.
[10] Wood NI, Glynn D, Morton AJ. “Brain training” improves cognitive performance and survival in a transgenic mouse model of Huntington’s disease. Neurobiol Dis. 2011;42(3):427-437.
[11] Zhang Y, Friedlander RM. Using non-coding small RNAs to develop therapies for Huntington’s disease. Gene Ther. 2011; 18(12):1139-1149.
[12] Laird CD. Proposed genetic basis of Huntington’s disease. Trends Genet. 1990;6(8):242-247.
[13] Harper PS, Morris MJ, Tyler A. Genetic testing for Huntington’s disease. BMJ. 1990;300(6732):1089-1090.
[14] Majid DS, Aron AR, Thompson W, et al. Basal ganglia atrophy in prodromal Huntington’s disease is detectable over one year using automated segmentation. Mov Disord. 2011;26(14):2544-2551.
[15] van den Bogaard SJ, Dumas EM, Acharya TP, et al. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. 2011;258(3):412-420.
[16] Nopoulos PC, Aylward EH, Ross CA, et al. Cerebral cortex structure in prodromal Huntington disease. Neurobiol Dis. 2010; 40(3): 544-554.
[17] Schneider SA, Bhatia KP. Huntington’s disease look-alikes. Handb Clin Neurol. 2011;100:101-112.
[18] Sułek-Piatkowska A, Krysa W, Zdzienicka E, et al. Searching for mutation in the JPH3, ATN1 and TBP genes in Polish patients suspected of Huntington’s disease and without mutation in the IT15 gene. Neurol Neurochir Pol. 2008;42(3):203-209.
[19] Nielsen TT, Mardosiene S, Løkkegaard A, et al. Severe and rapidly progressing cognitive phenotype in a SCA17-family with only marginally expanded CAG/CAA repeats in the TATA-box binding protein gene: a case report. BMC Neurol. 2012;12:73.
[20] Mori F, Tanji K, Toyoshima Y, et al. Optineurin immunoreactivity in neuronal nuclear inclusions of polyglutamine diseases (Huntington’s, DRPLA, SCA2, SCA3) and intranuclear inclusion body disease. Acta Neuropathol. 2012;123(5):747-749.
[21] Wilburn B, Rudnicki DD, Zhao J, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron. 2011; 70(3):427-440.
[22] Heng MY, Tallaksen-Greene SJ, Detloff PJ, et al. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27(34):8989-8998.
[23] Sack GH Jr. Mitochondrial matters in Huntington disease. J Bioenerg Biomembr. 2010;42(3):189-191.
[24] Ginestroni A, Battaglini M, Diciotti S, et al. Magnetization transfer MR imaging demonstrates degeneration of the subcortical and cortical gray matter in Huntington disease. AJNR Am J Neuroradiol. 2010;31(10):1807-1812.
[25] Sturrock A, Leavitt BR. The clinical and genetic features of Huntington disease. J Geriatr Psychiatry Neurol. 2010;23(4):243-259.
[26] Stout JC, Paulsen JS, Queller S, et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology. 2011;25(1):1-14.
[27] Huntington Study Group COHORT Investigators, Dorsey E. Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study. PLoS One. 2012;7(2):e29522.
[28] Ohta E. Clinical feature of neuroferritinopathy. Rinsho Shinkeigaku. 2012;52(11):951-954.
[29] Novak MJ, Tabrizi SJ. Huntington’s disease: clinical presentation and treatment. Int Rev Neurobiol. 2011;98:297-323.
[30] Raymond LA, André VM, Cepeda C, et al. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252-273.
[31] Estrada-Sánchez AM, Rebec GV. Role of cerebral cortex in the neuropathology of Huntington’s disease. Front Neural Circuits. 2013;7:19.
[32] Jones L, Hughes A. Pathogenic mechanisms in Huntington’s disease. Int Rev Neurobiol. 2011;98:373-418.
[33] Gusella JF, MacDonald ME. Hunting for Huntington’s disease. Mol Genet Med. 1993;3:139-158.
[34] Brinkman RR, Mezei MM, Theilmann J, et al. The likelihood of being affected with Huntington disease by a particular age, for a speci fi c CAG size. Am J Hum Genet. 1997;60(5):1202-1210.
[35] Langbehn DR, Hayden MR, Paulsen JS, et al. CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(2):397-408.
[36] Cao GN, Bao XH, Lu HM, et al. Clinical characteristics of Huntington disease in two pedigrees and analysis of expanded CAG trinucleotide repeat. Beijing Da Xue Xue Bao. 2011;43:163-167.
[37] Ha AD, Jankovic J. Exploring the correlates of intermediate CAG repeats in Huntington disease. Postgrad Med. 2011;123(5):116-121.
[38] Panegyres PK, Goh JG. The neurology and natural history of patients with indeterminate CAG repeat length mutations of the Huntington disease gene. J Neurol Sci. 2011;301(1-2):14-20.
[39] Klempíř J, Zidovská J, Stochl J, et al. The number of CAG repeats within the normal allele does not influence the age of onset in Huntington’s disease. Mov Disord. 2011;26(1):125-129.
[40] Brožová H, Stochl J, Klempíř J, et al. A sensitivity comparison of clinical tests for postural instability in patients with Huntington’s disease. Gait Posture. 2011;34(2):245-247.
[41] Vázquez-Mojena Y, Laguna-Salvia L, Laf fi ta-Mesa JM, et al. Genetic features of Huntington disease in Cuban population: Implications for phenotype, epidemiology and predictive testing. J Neurol Sci. 2013;335(1-2):101-104.
[42] Agostinho Lde A, Rocha CF, Medina-Acosta E, et al. Haplotype analysis of the CAG and CCG repeats in 21 Brazilian families with Huntington’s disease. J Hum Genet. 2012;57(12):796-803.
[43] Gatto E, Parisi V, Persi G, et al. Clinical and genetic characteristics in patients with Huntington’s Disease from Argentina. Parkinsonism Relat Disord. 2012;18(2):166-169.
[44] Baine FK, Kay C, Ketelaar ME, et al. Huntington disease in the South African population occurs on diverse and ethnically distinct genetic haplotypes. Eur J Hum Genet. 2013;21(10):1120-1127.
[45] Shannon KM. Huntington’s disease - clinical signs, symptoms, presymptomatic diagnosis, and diagnosis. Handb Clin Neurol. 2011;100:3-13.
[46] Imarisio S, Carmichael J, Korolchuk V, et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412(2):191-209.
[47] Dennhardt J, LeDoux MS. Huntington disease in a nonagenarian mistakenly diagnosed as normal pressure hydrocephalus. J Clin Neurosci. 2010;17(8):1066-1067.
[48] Trottier Y, Biancalana V, Mandel JL. Instability of CAG repeats in Huntington’s disease: relation to parental transmission and age of onset. J Med Genet. 1994;31(5):377-382.
[49] The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group. ACMG/ASHG statement. Laboratory guidelines for Huntington disease genetic testing. Am J Hum Genet. 1998;62(5):1243-1247.
[50] Raskin S, Allan N, Teive HA, et al. Huntington disease: DNA analysis in Brazilian population. Arq Neuropsiquiatr. 2000;58(4):977-985.
[51] Potter NT, Spector EB, Prior TW. Technical standards and guidelines for Huntington disease testing. Genet Med. 2004;6(1):61-65.
[52] Wild EJ, Mudanohwo EE, Sweeney MG, et al. Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008;23(5):716-720.
[53] Bech S, Petersen T, Nørremølle A, et al. Huntington’s disease-like and ataxia syndromes: identification of a family with a de novo SCA17/TBP mutation. Parkinsonism Relat Disord. 2010;16(1):12-15.
[54] Bodensteiner JB. A young man referred for an opinion regarding a neuroimaging abnormality. Semin Pediatr Neurol. 2010;17(1):45-48.
[55] Ohta M, Okuyama R, Ogawa E, et al. Cutaneous accumulation of abnormal polyglutamine proteins of patients with dentatorubralpallidoluysian atrophy. Eur J Neurol. 2009;16(11):1246-1249.
[56] Seixas AI, Holmes SE, Takeshima H, et al. Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Ann Neurol. 2012;71(2):245-257.
[57] Hire RR, Katrak SM, Vaidya S, et al. Spinocerebellar ataxia type 17 in Indian patients: two rare cases of homozygous expansions. Clin Genet. 2011;80(5):472-477.
[58] Suzuki K, Zhou J, Sato T, et al. DRPLA transgenic mouse substrains carrying single copy of full-length mutant human DRPLA gene with variable sizes of expanded CAG repeats exhibit CAG repeat length- and age-dependent changes in behavioral abnormalities and gene expression pro fi les. Neurobiol Dis. 2012;46(2):336-350.
[59] Ahmed Z, Tabrizi SJ, Li A, et al. A Huntington’s disease phenocopy characterized by pallido-nigro-luysian degeneration with brain iron accumulation and p62-positive glial inclusions. Neuropathol Appl Neurobiol. 2010;36(6):551-557.
[60] Panas M, Karadima G, Kalfakis N, et al. Co-segregation of Huntington disease and hereditary spastic paraplegia in 4 generations. Neurologist. 2011;17(4):211-212.
[61] Pillai JA, Hansen LA, Masliah E, et al. Clinical severity of Huntington’s disease does not always correlate with neuropathologic stage. Mov Disord. 2012;27(9):1099-1103.
[62] Rizk-Jackson A, Stoffers D, Sheldon S, et al. Evaluating imaging biomarkers for neurodegeneration in pre-symptomatic Huntington’s disease using machine learning techniques. Neuroimage. 2011;56(2):788-796.
Copyedited by James R, Zou LY, Li R, Yu J, Yang Y, Li CH, Song LP, Zhao M
10.4103/1673-5374.128258
Jiancheng Tu, M.D., Ph.D., Department of Clinical Laboratory Medicine & Center for Gene Diagnosis, Zhongnan Hospital of Wuhan University, Wuhan 430071, Hubei Province, China, 316099811@qq.com.
http://www.nrronline.org/
Accepted: 2013-12-20

- 中国神经再生研究(英文版)的其它文章
- Posterior quadrantic disconnection maintains the activity of isolated temporal-parietal-occipital nerve tissue: neuroprotective measures in the surgical treatment of epilepsy
- Circadian fl uctuations in three types of sensory modules in healthy subjects
- 7.0T nuclear magnetic resonance evaluation of the amyloid beta (1–40) animal model of Alzheimer’s disease: comparison of cytology veri fi cation
- Local inhibition of GABA affects precedence effect in the inferior colliculus
- Compound Formula Rehmannia alleviates levodopainduced dyskinesia in Parkinson’s disease
- The Pael-R gene does not mediate the changes in rotenone-induced Parkinson’s disease model cells