
重症多发性肌炎1例的诊治及文献复习
2014-03-20 08:50:31曲晨何慧薇冯美江鲁翔
实用老年医学 2014年2期
曲晨 何慧薇 冯美江 鲁翔
·病例报告·
重症多发性肌炎1例的诊治及文献复习
曲晨 何慧薇 冯美江 鲁翔
特发性炎症性肌病(idiopathic in⁃flammatorymyopathies,IIM)是一组以慢性肌肉炎症反应导致进行性肌萎缩为特点的系统性自身免疫病,包括多发性肌炎(polymyositis,PM)、皮肌炎(dermatomyo⁃sitis,DM)和包涵体肌炎(inclusior body myositis,IBM)等。其中PM是一组病因不清的弥漫性肌肉炎症性疾病,主要临床症状为对称性四肢近端、颈肌、咽部肌肉无力,肌肉压痛,血清酶增高及肌电图的异常,常见骨骼肌无力、肌痛,继之产生肌肉萎缩。而本例患者出现除骨骼肌外,心、肺、消化道等肌群均有受累的表现,病情凶险,发展迅速,危及生命,同时也给诊断带来困难。目前重症PM的病例国内外鲜有报道,现将其诊疗经验及文献复习报道如下。
1 病例资料
1.1 一般情况 患者女性,79岁,因“四肢酸痛1年,腰背部疼痛伴下肢水肿1周”入院,既往有脑梗死史,高血压病史,否认有他汀类药物使用史,否认有小龙虾食用史,家族中有PM病史。患者1年前出现四肢酸痛,未引起重视,1周前无明显诱因下出现腰背部疼痛,后进行性加重,伴有四肢无力,疼痛,水肿。病程中患者颜面及四肢浮肿,感颈部无力,吞咽困难,偶有呛咳,饮食差,二便正常,入院查体血压110/60 mmHg,消瘦,端坐呼吸,未及明显皮疹,浅表淋巴结未及肿大,两肺呼吸音粗,两下肺布满湿啰音,心率100次/min,律齐,可及舒张期奔马律。腹平软,Murphy征可疑阳性,右肾区叩击痛,左上肢及双下肢Ⅲ°可凹性水肿。四肢肌力1级,病理反射未引出。
1.2 实验室检查 丙氨酸氨基转移酶88 IU/L,天冬氨酸氨基转移酶153 IU/L,磷酸肌酸激酶2308 IU/L,乳酸脱氢酶1040 IU/L,血沉24mm/h,血清肌红蛋白2113.00 ng/ml,血清肌酸激酶同工酶(CK⁃MB)58.28 ng/ml,血清肌钙蛋白T 0.389 mg/m l。脑钠钛>10 000,乙肝两对半均为阴性,免疫球蛋白IgA 3.96 g/L,IgG 17.30 g/L,IgM 2.57 g/L,SSA抗体阳性,抗核抗体阳性,Jo⁃1抗体阳性。CT(图1)示两下肺炎,左下肺肺不张,两肺胸腔积液,两肺多发肺大疱。肌电图示肌源性损害。
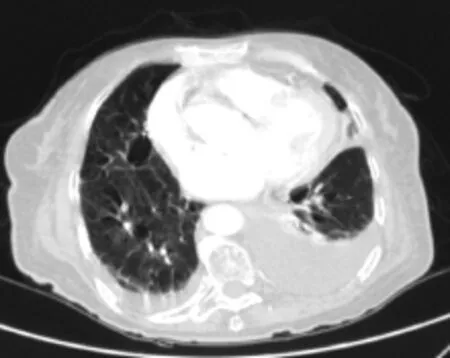
图1 CT表现
1.3 治疗 患者入院后予以激素冲击治疗,心肌酶谱进行性升高,心电图为下壁缺血性改变,伴有室性早搏。急诊冠脉CT未见明显异常。考虑PM伴有心肌损害,患者心衰症状明显,予以利尿扩冠。后患者出现肌力减退,呼吸肌麻痹,呼吸衰竭,意识不清,不能自主进食,予以无创通气,抗感染,鼻饲饮食。并加用丙种球蛋白支持治疗。2周后患者呼吸功能改善,去除呼吸机,四肢肌力2级。4周后患者肌力5级,可自行进食,加用环磷酰胺。3月后患者生活基本可以自理,复查酶谱正常。随访至今。
2 讨论
2.1 病因及流行病学 目前认为PM与自身免疫紊乱有关。也有部分学者认为与病毒感染(特别是副黏液病毒,如柯萨奇A9病毒)或遗传因素有关[1]。多为亚急性起病,任何年龄均可发病,在儿童5~14岁和成人45~60岁各出现一个高峰,文献报道PM的男女比例分别为1∶5,发病率约每年(2~5)/10万,部分患者存在恶性肿瘤,约20%患者合并红斑狼疮、硬皮病、类风湿性关节炎等其他自免疫性疾病[2]。当累及心肌细胞时,致心内膜增生、中膜坏死、侵及传导系统的心肌细胞时引起不同种类的心律失常[3]。而本例患者无明显的诱因及家族遗传史。
2.2 临床症状 PM特征性表现为双侧对称性近端肌无力,肩胛带及骨盆带肌常受累,进行性对称性红斑角皮症和眶周及内眦处紫红色皮疹,常伴眶周水肿及上睑毛细血管扩张。紫红色皮疹还可出现于颈前上胸部(V区)及颈后上肩背部(披肩征)。常见并发症为间质性肺炎。本例患者多个肌群均受累[4],其颈肌受累表现为卧位时抬头困难,坐位时无力仰头。咽喉肌受累表现为吞咽困难、饮水呛咳等。呼吸肌受累表现为呼吸困难,使用无创呼吸机辅助通气。心肌受累表现为胸闷,急性心力衰竭。消化道肌群受累表现为反酸、腹胀等症状。
2.3 实验室检查 PM中炎细胞主要为CD8+T细胞。血清肌酶谱检查有较高的敏感性,血清肌酶谱检查不仅具有诊断价值,而且能反映病情轻重和治疗情况(其反映肌纤维坏死程度)。本例患者肌酸激酶(CK)和乳酸脱氢酶明显升高,激素治疗数周后CK水平明显下降,且出现在肌力改善之前,然后吞咽困难有改善。抗Jo⁃1抗体是诊断本病的标志性抗体,阳性率为25%,在合并有肺间质病变的患者中其阳性率可达60%。ANA阳性率为20%~30%[5],对肌炎诊断不具有特异性。ANA滴度的高低与疾病的活动性相关,可以作为治疗中监测效果的指标之一。肌电图检查显示为肌源性损害。肌肉活检可见肌细胞受损坏死、炎细胞浸润。本例患者还存在CK⁃MB,TNI升高,提示患者并发了心肌损害。应及时与急性心肌梗死相鉴别,结合患者ECG改变和冠脉CT结果容易判断是否为急性心肌梗死。急性心肌梗死时,心肌标志物升高存在动态演变的规律,该患者心肌标志物无动态演变,ECG亦无动态变化,超声心动图未见明确室壁节段运动异常,均不支持急性心肌梗死。
2.4 诊断标准 1975年Bohan等[6]提出PM诊断标准:(1)肢带肌、颈前肌对称无力,病程持续数周到数月,有/无吞咽困难、呼吸肌受累。(2)肌肉活检:肌纤维坏死,炎细胞浸润,束周肌萎缩。(3)血清CK升高。(4)肌电图:肌源性损害,皮肤改变。具备4项者可确诊为,具备3项者可能为PM,只具备2项者为疑诊PM。1991年Dalakas等[7]提出以肌肉活检结果为主的新标准。2003年Dalakas提出CD8/MHC⁃Ñ复合物是PM和IBM特异性标志,建议将其引入PM诊断标准中。但至今仍沿用1975年Bohan和Peter的诊断标准。
2.5 治疗 PM的治疗包括糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白及血浆置换。而特异性的免疫靶向治疗尚在研究中。糖皮质激素是治疗的首选药。患者入院后前3日使用激素冲击治疗,然后强的松60 mg/d。维持4周后减量。维持用药2年左右。类固醇药物引起肌无力可能加重心衰及呼衰,应积极对症治疗。出现呼衰且合并肺部感染的患者,应尽早使用胃管,避免因误吸致吸入性肺炎。由于患者咽反射减退,呼吸肌麻痹者应早行无创通气或气管切开,必要时采用机械通气,并给予肠外营养支持。患者合并有心肌损害时,应动态观察心肌酶谱及心电图的变化,可鉴别是否合并急性心肌梗死,急诊冠脉CT或PCI也可鉴别。对于重症PM或者激素治疗效果差时可使用丙种球蛋白,改善肌无力症状,且其耐受性良好。免疫抑制剂常加用甲氨蝶呤以减少激素用量,也可加用硫唑嘌呤[8],但本例患者考虑合并有肺部感染,呼吸衰竭,所以在感染控制后加用环磷酰胺。根据美国呼吸学会推荐合并有间质性肺炎的患者使用激素联合环磷酰胺或硫唑嘌呤治疗[9⁃10]。
总之对于重症PM,除了及早足量使用激素外,应重视心肺功能的支持与恢复,避免严重的并发症,必要时加用免疫抑制剂及免疫球蛋白,从而降低死亡率。
[1] Yu KH,Wu YJ,Kuo CF,et al. Survival analysis of patientswith der⁃matomyositis and polymyositis:analysis of 192 Chinese cases[J]. Clin Rheumatol,2011,30(12):1595⁃1601.
[2] 蔡云雅,方红.恶性肿瘤相关性皮肌炎/多发性肌炎[J].国际皮肤性病学杂志,2012,38(1):51⁃54.
[3] Bazzani C,Cavazzana I,Ceribelli A,et al.Cardiological features in idio⁃pathic inflammatory myopathies[J]. J Cardiovasc Med(Hagerstown),2010,11(12):906⁃911.
[4] Baer AN.Differential diagnosis of idi⁃opathic inflammatory myopathies[J]. Cur Rheumatol Rep,2006,8(3):178⁃187.
[5] 宋晓颖,王淑义.多发性肌炎/皮肌炎研究现状[J].实用儿科临床杂志,2007,22(9):704⁃706.
[6] Bohan A,Peter JB.Polymyositis and dermatomyositis(first of two parts)[J].N Engl JMed,1975,292(7):344⁃347.
[7] Dalakas MC.Polymyositis,dermato⁃myositis and inclusion⁃body myositis[J].N Engl JMed,1991,325(21):1487⁃1498.
[8] 王亚慧,苏振丽.老年人重症肌无力临床研究[J].实用老年医学,2010,24(4):297⁃299.
[9] 罗志兵,沈策.多发性肌炎/皮肌炎肺间质病变的治疗及预后[J].临床肺科杂志,2008,13(12):1637⁃1638.
[10]施举红,许文兵,刘鸿瑞,等.多发性肌炎皮肌炎合并肺间质性病变的临床特征[J].中华结核和呼吸杂志,2008,31(4):250⁃254.
R 593
B
10.3969/j.issn.1003⁃9198.2014.02.027
2013⁃06⁃22)
210011江苏省南京市,南京医科大学第二附属医院老年科
猜你喜欢
现代临床医学(2022年2期)2022-04-19 12:48:52
中国民间疗法(2021年17期)2021-11-04 08:39:30
中华养生保健(2020年7期)2020-11-16 01:14:28
家庭医学(下半月)(2019年11期)2020-01-16 08:39:12
高中生·天天向上(2017年4期)2017-06-09 02:37:17
反射疗法与康复医学(2017年7期)2017-01-16 01:11:02
中国药物应用与监测(2015年5期)2015-12-11 03:15:57
中国医疗美容(2015年1期)2015-07-12 10:06:04
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
疑难病杂志(2014年12期)2014-04-16 05:19:30
