
两步法合成四种芳基苄基酮
2014-03-14 05:56李艺林徐崇福邓云香朱建华郑黄利王颖
精细石油化工 2014年6期
李艺林,徐崇福,邓云香,朱建华,郑黄利,王颖
(常州大学石油化工学院,江苏 常州 213164)
芳基苄基酮是一类重要的医药中间体,可用于合成环氧合酶抑制剂[1-2]。目前,在已报道的芳基苄基酮合成中,首先要得到相应的芳基苄基醇先驱体,主要方法有:1)以4-氯苯甲醛,氯化苄为原料,在Zn/CdCl2/InCl3催化下反应3h,产率为64%[3];2)以4-氯苯甲醛、氯化苄为原料,在K2HPO4/CH3OH以及Zn和AgNO3组成的复合催化体系中反应得到4-氯苯基苄基醇,产率为60%[4];3)利用微波辐射,使苄基溴化锌与带吸电子取代基的芳香醛在Pd(PPhs)2C12催化下发生加成反应,得到仲醇,产率为47%[5]。但是,这些方法都需要贵重金属催化剂,成本较高,而且产物复杂,分离纯化困难较大。
笔者以氯化苄为起始原料,与金属镁反应原位生成的苄基氯化镁与芳基甲醛进行亲核加成,质子化脱镁合成一系列芳基基苄基醇,进一步氧化脱水得到相应的芳基苄基酮。该法具有原料便宜易得,反应简单,副反应少,收率高等优点。合成路线如下所示。

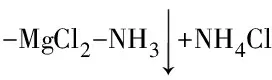
1 实验部分
1.1 原料与仪器
镁条、氯化苄、对氯苯甲醛等均为市售AR。无水乙醚溶剂须通过去水装置,使用金属钠除水;二苯甲酮为指示剂,加热回流至溶剂呈蓝色时,蒸出使用。
瑞士Broker 500 MHz超导核磁仪,瑞士Broker 400 MHz超导核磁仪,德国Bruker TENSOR27原位红外仪。
1.2 芳基苄基甲醇的合成
1.2.1苯基苄基甲醇的合成
在置有搅拌器、冷凝管和恒压滴液漏斗的250 mL四口烧瓶(全程氩气保护)中加入0.53 g(0.022 mol)金属镁条,15 mL无水乙醚和一粒碘晶体。在恒压滴液漏斗中加入2.53 g(0.020 mol)氯化苄,用15 mL无水乙醚稀释成溶液。首先向烧瓶中放入约1/10体积的氯化苄乙醚混合液,并将其下置于温水浴升温引发反应。在搅拌下将剩余的混合液适速滴加至四口烧瓶中,反应温度控制在30 ℃,滴加完毕后继续搅拌1.0 h,使所有氯化苄消耗殆尽。然后改换冰水浴将格氏反应混合物冷却至5 ℃左右,并缓慢滴加由1.38 g(0.013 mol)苯甲醛和20 mL无水乙醚组成的混合液。
投料完成后,逐渐升温总反应母液,连续回流2.0 h。用冰水浴将总反应母液冷却至5 ℃左右,滴加饱和氯化铵水溶液进行水解。过滤出去残余金属镁和机械杂质。将滤液转移至分液漏斗中静置分层,分离并收集有机相,水层用乙醚萃取两次,将萃取液与有机相合并,弃去水相。有机相用无水硫酸镁干燥2.0 h,减压旋蒸除去溶剂得到3.39 g油状粗产物,从中称取0.326 g试样进行柱色谱分离(流动相为V(石油醚)∶V(乙酸乙酯)=15∶1),减压旋蒸得白色固体0.211 g纯品,分离产率85.1%。
1H NMR(500 MHz,CDCl3),δ:1.95(s,1H,OH),3.00~3.06(m,2H,CH2),4.88~4.91(m,1H,CHO),7.19~7.37(m,10H,Ph);13C NMR(125 MHz,CDCl3),δ:46.06(CH2),75.32(CHO),125.89(Ph),126.59(Ph),127.59(Ph),128.44(Ph),129.51(Ph),138.04(Ph),143.80(Ph)。
IR(KBr),σ/cm-1:3 315(O—H),3 026~2 863(C—H),1 601~1 445(CC),1 072,1 041,742,697(finger)。
1.2.24-氯苯基苄基甲醇的合成
操作方法同1.2.1。试剂用量:0.97 g(0.04 mol)金属镁条,4.43 g(0.035 mol)氯化苄;3.51 g(0.025 mol)4-氯苯甲醛。将母液有机相减压旋蒸得到油状粗产物7.54 g,从中称取0.377 g试样进行柱色谱分离,减压旋蒸得到白色固体0.255 g纯样,分离产率87.7%。
1H NMR(500 MHz,CDCl3),δ:1.96~1.97(m,1H,OH),2.93~3.03(m,2H,CH2),4.86~4.90(m,1H,CHO),6.86~6.89(m,2H,Ph),7.17~7.32(m,7H,Ph);13C NMR(125 MHz,CDCl3),δ:46.05(CH2),74.64(CHO),127.02(Ph),127.34(Ph),128.60(Ph),129.49(Ph),133.18(Ph),137.51(Ph),142.19(Ph)。
IR(KBr),σ/cm-1:3 370(O—H),2 923~2 858(C—H),1 597~1 494(CC), 1 088,1 030(finger)。
1.2.34-甲氧基苯基苄基甲醇的合成
操作方法同1.2.1。试剂用量:0.97 g(0.04 mol)金属镁条,4.43 g(0.035 mol)氯化苄;3.40 g(0.025 mol)4-氯苯甲醛。将母液有机相减压旋蒸得到油状粗产物7.44 g,从中称取0.372 g试样进行柱色谱分离,减压旋蒸得到白色固体0.236 g纯样,分离产率81.9%。
1H NMR(500 MHz,CDCl3),δ:1.92~1.93(m,1H,OH),2.96~3.02(m,2H,CH2),3.80(s,3H,OCH3),4.83~4.86(m,1H,CHO),6.86~6.89(m,2H,Ph),7.17~7.31(m,7H,Ph);13C NMR(125 MHz,CDCl3),δ:45.26(CH2),55.45(CHO),113.79(Ph),126.77(Ph),128.64(Ph),129.38(Ph),129.64(Ph),130.91(Ph),130.94(Ph),134.98(Ph)。
IR(KBr),σ/cm-1:3 279(O—H),2 936~2 879(C—H), 1 514~1 454(CC),1 251,1 174,1 034,832,699(finger)。
1.2.41,1′-对苯基二苄基二甲醇的合成
操作方法同1.2.1,但在格氏反应混合液与对苯二甲醛的反应中使用无水四氢呋喃代替无水乙醚以便提高溶解性。试剂用量:0.97 g(0.04 mol)金属镁条,4.43 g(0.035 mol)氯化苄,2.01 g(0.015 mol)对苯二甲醛。将母液有机相减压旋蒸得到油状粗产物5.76 g,在无水乙醇中重结晶得到白色固体2.14 g纯样,分离产率44.9%。
1H NMR(400 MHz,CDCl3),δ:1.951~1.956(m,2H,2xOH),2.961~3.061(m,4H,2xCH2),4.887~4.918(m,2H,2xCHO),7.183~7.334(m,10H,Ph);13C NMR(100 MHz,CDCl3),δ:45.49(CH2),126.88(Ph),128.60(Ph),128.69(Ph),129.43(Ph),129.46(Ph),133.15(Ph),134.54(Ph),136.60(Ph)。
IR(KBr),σ/cm-1:3 382~3 319(O—H),3 086~2 905(C—H),1 496~1 417,1 050(CC),875,854,823,696(finger)。
1.3 芳基苄基酮的合成
1.3.1苯基苄基酮的合成
在装有恒压滴液漏斗、回流冷凝器和机械搅拌装置的100 mL三口烧瓶中,加入3.96 g(0.02 mol)苯基苄基甲醇和20 mL二氯甲烷,缓缓加入1.5 g氯铬酸吡啶硅胶复合物(PCC/硅胶)。开启搅拌,在30 ℃温水浴下反应2.0 h,使用薄层色谱法监测反应进程直到反应完成。过滤除去固体残渣。用50 mL蒸馏水分次洗涤滤液3次,用分液漏斗分出有机相,用20 mL二氯甲烷将水相萃取两次,合并有机相。将有机溶液减压旋蒸得到油状粗产物,在50 mL乙醇中重结晶得到白色固体,干燥后获纯样3.28 g,分离产率83.6%。
1H NMR(400 MHz,CDCl3),δ:4.287(s,2H,CH2),7.235~7.341(m,5H,Ph),7.441~7.569(m,3H,Ph),8.005~8.022(m,2H,Ph);13C NMR(100 MHz,CDCl3),δ:46.06(CH2),75.10(CHO),125.98(Ph),126.64(Ph),128.51(Ph),129.52(Ph),137.93(Ph),143.177(CO)。
IR(KBr),σ/cm-1:3 100~2900(C—H),1 686(CO),1 578(CC),1 218,1 201(finger)。
1.3.24-氯苯基苄基酮的合成
操作方法同1.3.1。试剂用量:4.65 g(0.02 mol)4-氯苯基苄基醇,1.5 gPCC/硅胶,得到白色晶体纯样3.94 g,分离产率85.5%。
1H NMR(500 MHz,CDCl3),δ:4.26(s,2H,CH2),7.24~7.43((m,7H,Ph),7.94~7.95(m,2H,Ph);13C NMR(125 MHz,CDCl3),δ:45.52(CH2),127.03(Ph),128.76(Ph),128.95(Ph),129.37(Ph),130.03(Ph),134.14(Ph),134.79(Ph),139.60(Ph),196.48(CO)。
IR(KBr),σ/cm-1:3 100~2 900(C—H),1 687(CO),1 588(CC),1 089,1 072(finger);。
m.p.107.9~108.6 ℃。
1.3.34-甲氧基苯基苄基酮的合成
操作方法同1.3.1。试剂用量:4.56 g(0.02 mol)4-甲氧基苯基苄基甲醇,1.5 gPCC/硅胶,得到白色晶体纯样3.63 g,分离产率80.4%。
1H NMR(500 MHz,CDCl3),δ:3.86(s,3H,CH3),4.23(s,2H,CH2),6.91~6.93(m,2H,Ph),7.22~7.33(m,5H,Ph),7.99~8.00(m,2H,Ph);13C NMR(125 MHz,CDCl3),δ:45.99(CH2),55.27(CH3O),113.76(Ph),126.52(Ph),127.16(Ph),128.45(Ph),129.47(Ph),129.50(Ph),136.01(Ph),138.17(Ph),159.05(CO)。
IR(KBr),σ/cm-1:3 100~2 900(C—H),1 673(CO),1 575(CC),1 110,1 074(finger)。
m.p. 73.5~74.5 ℃。
1.3.41,1′-对苯基二苄基二酮的合成
操作方法同1.3.1。试剂用量:4.77 g(0.015 mol)1,1′-对苯基二苄基二甲醇,1.5 gPCC/硅胶,得到白色固体纯样3.65 g,分离产率77.5%。
1H NMR(400 MHz,CDCl3),δ:4.294(s,4H,2xCH2),7.242~7.345(m,10H,Ph),8.059(s,4H,Ph);13C NMR(100 MHz,CDCl3),δ:45.874(CH2),127.115(Ph),128.803(Ph),128.827(Ph),129.415(Ph),133.924(Ph),139.733(Ph),197.02(CO)。
IR(KBr),σ/cm-1:3 100~2 900(C—H),1 685(CO),1 498(CC),1 218,989(finger)。
m.p.178.8~179.9 ℃。
2 结果与讨论
2.1 芳基苄基甲醇的工艺条件
在无水条件下,氯苄与新鲜金属镁削首先在相界面发生氧化还原反应,原位生成苄基氯化镁进入液相与适时引入的芳基甲醛进行亲核加成反应,分别在芳基甲醛的羰基碳原子和氧原子上引入苄基和氯化镁基团,再与氯化铵的脱氨氢解即可得到芳基苄基甲醇。因为苄基氯化镁(格氏试剂)具有高活性,它有可能通过自由基通道自身发生苄基偶联和分子歧化,反应机理见图1。
以苯基苄基甲醇的合成为例,对原位合成苄基氯化镁以及后续与苯甲醛亲核加成反应的工艺条件进行了考察。

图1 芳基苄基甲醇的形成机理
2.1.1镁的物理状态的影响
表1是镁的物理状态的影响。

表1 镁的物理状态的影响
从表1可以看出:未经活化的镁粉制备苄基氯化镁时,反应引发较难,导致苯基苄基甲醇产率很低;经活化处理的镁粉,产率有所提高;而新鲜镁带锉出的镁粉反应极易引发,产率达到85.1%。这表明使用未受到空气氧化作用的新鲜镁屑有利于提高产物产率。
2.1.2苄基氯化镁制备温度的影响
苄基氯化镁制备温度对产率的影响见表2。从表2可以看出:苄基氯化镁的原位生成须要在合适的反应温度下进行。低温时,很少分子达到发生反应所需的最低活化能,结果是目标产物产率较低。随着温度提高,目标产物产率迅速提高;超过30 ℃时,产率反而降低,这是由于当温度接近乙醚沸点时,大量溶剂进入气相不利于反应物的分子接触。另外,活泼的氯化苄在高温下易与苄基氯化镁进行偶联反应,生成1,2-二苯乙烷导致产率下降。

表2 苄基氯化镁制备温度的影响
2.1.3反应物投料比的影响
表3为反应物投料比的影响。由表3可见,随着苯甲醛加入量的增加,产率下降。这是因为原位生成的格氏试剂苄基氯化镁并没有按照化学计量生成,大约只有理论值的65%。因此按理论值加入等当量苯甲醛不能完全反应,既浪费试剂又造成产物分离困难。根据实验结果确定n(镁)∶n(氯化苄)∶n(苯甲醛)= 1.10∶1.00∶0.65。在计算苯基苄基甲醇产率时,以苯甲醛作为控制试剂。
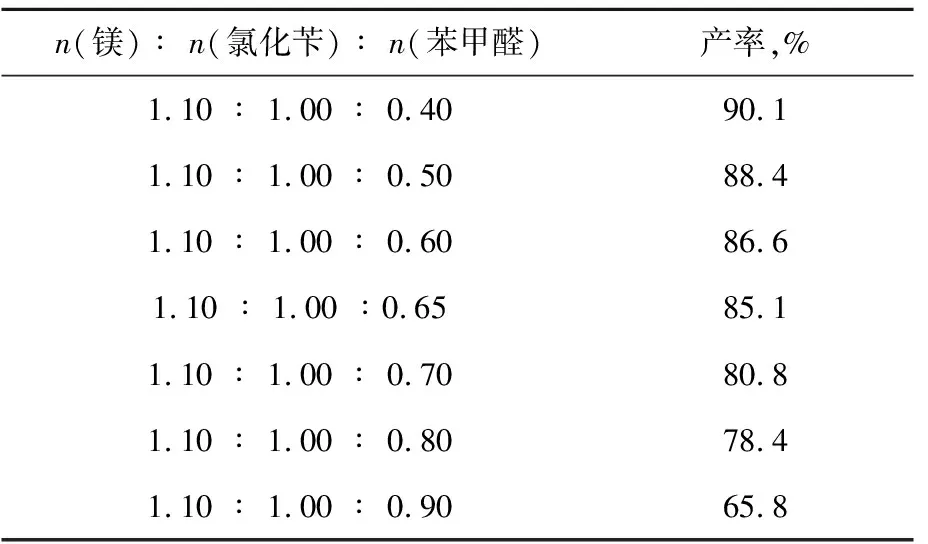
表3 反应物投料比的影响
2.1.4镁与氯化苄反应时间的影响
表4为镁与氯化苄反应时间的影响。从表4中看出:起初反应产率随时间增加而增加。当反应时间超过1.5 h的时候,反应的产率不仅没有增加反而下降了。由此判断,原位生成的苄基氯化镁浓度在1 h达到最大值。如果不及时引入苯甲醛,格氏试剂本身就会通过自由基反应通道发生偶联副反应,因此镁和氯化苄的反应时间应该控制在1 h。
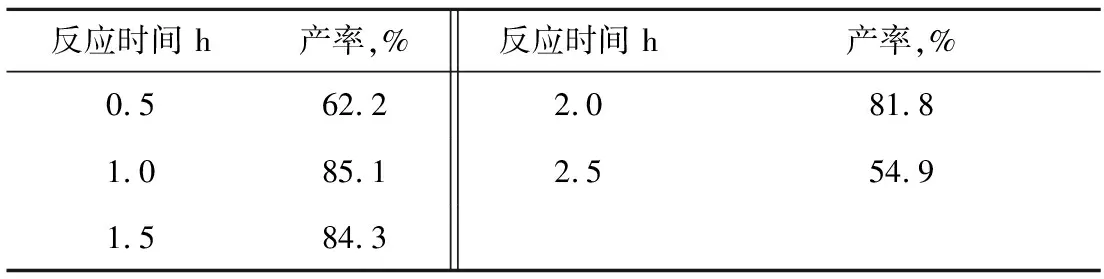
表4 镁与氯化苄反应时间对苯基苄基甲醇收率的影响
2.1.5苄基氯化镁与苯甲醛反应时间的影响
表5是苄基氯化镁与苯甲醛反应时间的影响。
由表5可见:随着反应时间增加,苯基苄基甲醇的产率逐步升高。表明在有苯甲醛存在下,苄基氯化镁与苯甲醛之间的亲核加成反应有效地抑制了两个苄基氯化镁之间的自由基偶合反应的发生。综合考虑,反应时间选择在2.0 h。
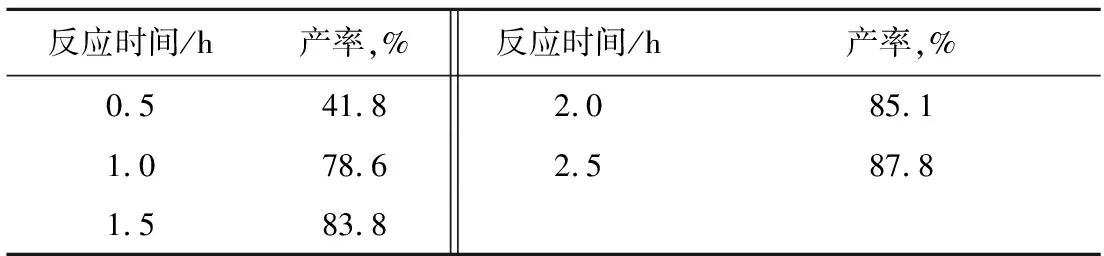
表5 苄基氯化镁与苯甲醛反应时间的影响
2.2 氯铬酸吡啶盐催化氧化法合成芳基苄基酮的工艺条件
用氯铬酸吡啶盐催化氧化芳基苄基甲醇,得到相应的芳基苄基酮。氯铬酸吡啶盐在该反应体系中起着酸碱双重作用。首先,这个复合盐的解离产生游离的氯铬酸和吡啶。氯铬酸与芳基苄基甲醇进行酯化反应脱去一分子水生成氯铬酸芳基苄基甲酯,吡啶对于α-氢的亲核偶合产生吡啶质子复合体,氯化二氧化铬负离子,并释放出目标产物芳基苄基酮。金属铬最终的归宿将是正三价离子。由芳基苄基甲醇催化氧化合成芳基苄基酮的反应机理如图2所示。
以氯铬酸吡啶盐催化氧化苯基苄基甲醇为先行体合成苯基苄基酮为例,考察了芳基苄基酮的工艺条件。
2.2.1反应温度的影响
表6是反应温度的影响。由表6可见:在5~25 ℃时,苯基苄基酮产率随着温度的升高而增加。继续升高反应温度,产率反而下降;这可能是在高温时,产物苯基苄基酮进一步被深度氧化,副产物增加。
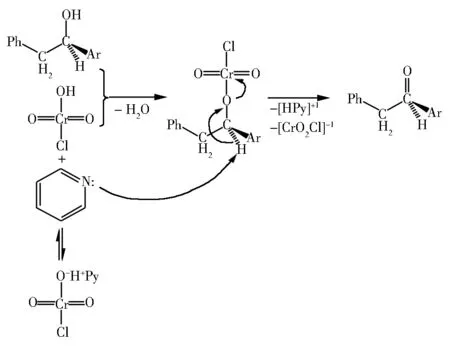
图2 氯铬酸吡啶盐催化氧化芳基苄基甲醇的反应机理

反应温度/℃产率,%反应温度/℃产率,%521.32583.61554.23575.9
2.2.2反应时间的影响
表7是反应时间的影响。

表7 反应时间的影响
由表7可见:在2.0 h之前,收率随着反应时间的增加而增加,当时间达到2.0 h时达到最大值。2.0 h后,可能是因为过度氧化,导致苯基苄基酮产率下降。
2.2.3氧化剂用量的影响
表8是氧化剂用量的影响。由表8可见:当PCC/硅胶用量为1.5 g时,这时收率最高达到83.6%,当再增加PCC/硅胶用量时,产率略有下降,这可能是因为过度氧化,部分产物可能被摧毁所致。

表8 氯铬酸吡啶硅胶复合体用量的影响
3 结 论
a.以氯化苄和对位取代的苯甲醛为原料,通过格氏法合成了芳基苄基甲醇。将这些化合物进一步经硅胶负载氯铬酸吡啶(PCC)氧化,得到相应的苯基苄基酮。所有产物都得到分离提纯,它们的分子结构经1H,13C核磁波谱及其红外波谱表征。
b.反应使用新鲜锉出的镁条来制备格式试剂,氧化过程中也使用了氯铬酸吡啶盐催化,与已报道的需要使用贵金属催化剂的合成方法相比,原料便宜易得。反应是用格式试剂与醛酮类反应,反应全程温度0~30 ℃,时间2~3 h,条件温和且时间较短。
c.文献[6]中只考察了一种基团,本文考察在多种不同取代基团下的可行性,并对其工艺条件进行了优化,总产率在70%以上。其中在以对苯二甲醛为原料的反应条件优化过程中,发现使用乙醚作为溶剂基本没有产物,而使用四氢呋喃作为溶剂够取得较好的产率。
参 考 文 献
[1] Venugopal R V, Manojit P. A high speed parallel synthesis of 1,2-diaryl-1-ethanones via a clean-chemistry C—C bond formation reaction [J]. Tetrahedron, 2003: 3283-3290.
[2] Lila Karimi, Latifeh Navidpour. Syntheses of 4,5-Diaryl-1,2,3-thiadiazoles [J]. Phosphorus, Sulfur Silicon Relat Elem, 2005, 180:1593-1600.
[3] Zhou Cunliu, Jiang Jiaoyang, Zhou Yuqing, et al. Chemoselective carbonyl benzylation mediated by Zn/CdCl2/InCl3in tap water. Tetrahedron Lett, 2005, 2: 61-64.
[4] Lothar W, Bieber, Elisabeth C. et al. Silver catalyzed zinc Barbier reaction of benzylic halides in water [J]. Tetrahedron Lett, 1998, 39: 9393-9396.
[5] 张玉梅.有机锌试剂与羰基化合物的环境友好化学反应研究[D].西北师范大学:学位论文,2005,100-108.
[6] 王昕宇,徐崇福,陈颖,等.药物中间体4-氯苯基苄基酮的合成[J].常州大学学报:自然科学版,2012,24(2):31-34.
猜你喜欢
盐科学与化工(2022年5期)2022-05-19
天津化工(2020年2期)2020-05-09
盐科学与化工(2020年2期)2020-03-19
合成化学(2015年2期)2016-01-17
合成化学(2015年10期)2016-01-17
合成化学(2015年1期)2016-01-17
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
中国科技纵横(2015年17期)2015-09-19
中国洗涤用品工业(2015年9期)2015-02-28
