
微波辅助合成半乳聚糖及其结构分析
2014-03-08 09:17王海松施用晖乐国伟
食品科学 2014年24期
王海松,施用晖,2,乐国伟,2,*
(1.江南大学食品学院,食品营养与功能因子研究中心,江苏 无锡 214122;2.江南大学 食品科学与技术国家重点实验室,江苏 无锡 214122)
微波辅助合成半乳聚糖及其结构分析
王海松1,施用晖1,2,乐国伟1,2,*
(1.江南大学食品学院,食品营养与功能因子研究中心,江苏 无锡 214122;2.江南大学 食品科学与技术国家重点实验室,江苏 无锡 214122)
目的:研究半乳聚糖的快速、高效制备方法,并对产物进行结构分析。方法:在微波辐照下,以半乳糖为底物,氯化钠溶液为引发剂,杂多酸为反应催化剂,催化底物脱水缩合生成半乳聚糖,并通过乙醇沉淀去除催化剂和引发剂,Sephadex G-25葡聚糖凝胶柱分级纯化,高效凝胶渗透色谱分析半乳聚糖的纯度及聚合度,高效阴离子交换色谱分析单糖组成,最后通过红外光谱分析半乳聚糖的构型。结果:半乳聚糖的最优合成条件为引发剂浓度0.25 mol/L、催化剂添加量1.1%、反应温度130 ℃、微波辐照4.5 min,半乳糖转化率为97.22%。半乳聚糖的平均分子质量为2.853 kD,平均聚合度为17。单糖组成为半乳糖及微量葡萄糖。红外光谱及氢核磁共振波谱(1H-nuclear magnetic resonance,1H-NMR)分析表明半乳聚糖残基以β-构型为主。结论:可以通过微波辐照杂多酸催化的方法快速、高效制备半乳聚糖。
微波;半乳聚糖;结构分析
半乳聚糖是指由半乳糖构成或以半乳糖为主、其他单糖为辅构成的直连或支链聚合物及其衍生物的统称[1]。半乳聚糖具有免疫活性[2]、抗病毒活性[3-4]、抗凝血活性[5]以及抑制肿瘤转移[6]等功能,并且可作为膳食纤维用于食品加工[7]。
目前,半乳聚糖主要是通过海洋藻类(如红藻、褐藻、角叉菜胶等)或陆生植物(如刺槐、马尾松、苍术、当归)及动物组织提取,然后通过水解及衍生化等方法获得[1],制备工艺繁琐、复杂。因此,快速、有效的制备方法为人们所期待。微波在多糖提取方面多有应用[8],其是否可用于多糖的合成,尚待研究。微波辅助有机合成作为一种快速、有效的合成手段已有广泛研究[9]。王海松等[10]曾报道了微波辅助快速合成低聚糖的方法。基于糖在酸催化作用下加热可脱水形成聚合物[11-12],若能够控制产物的聚合度,那么,采用微波辅助合成多糖将成为可能。
本实验在微波辅助合成低聚糖的基础上,改进合成条件,以半乳糖为原料,对微波辅助合成半乳聚糖的条件进行探索,并对产物结构进行解析。
1 材料与方法
1.1 材料与试剂
D-半乳糖(Gal)、氯化钠(NaCl)、三氟乙酸、无水乙醇(99%) 国药集团化学试剂有限公司;杂多酸江南大学食品营养与功能因子研究中心;Sephadex G-25葡聚糖凝胶 北京索莱宝科技有限公司。
1.2 仪器与设备
XH-200A型电脑微波固液相合成/萃取工作站 北京祥鹄科技发展有限公司;BSZ-100型自动部分收集器 上海青浦沪西仪器厂;1260型高效液相色谱仪(配Shodex RI-101视差检测器) 美国Agilent公司;ICS-5000型离子色谱仪器 美国Dionex公司;600型高效凝胶渗透色谱仪(配2410视差检测器) 美国Waters公司;560傅里叶变换红外光谱仪 美国Nicolet公司;Avance III-400MHz核磁共振仪 德国Bruker公司。
1.3 方法
1.3.1 半乳聚糖合成的单因素试验
烧杯中加入25 g半乳糖,然后添加1.1%杂多酸作为催化剂,3.75 mL 0.3 mol/L氯化钠溶液作为反应引发剂,三者充分搅拌,混匀后送入微波反应器,并固定烧杯。设定微波功率800 W、反应温度130 ℃、反应时间5 min,开启搅拌器120 r/min,并开启微波合成仪,反应结束后,测定半乳糖的转化率。固定底物添加量,引发剂添加量及微波功率,分别考察反应温度(100~150 ℃)、催化剂添加量(0.5%~1.5%)、微波辐照时间(4~6 min)、引发剂浓度(0.1~0.35 mol/L)对半乳糖转化率的影响。
1.3.2 正交试验设计
以半乳糖的转化率为指标,以反应温度、微波辐照时间、引发剂浓度、催化剂添加量为影响因素,根据单因素试验结果,选取适宜的因素水平进行L16(44)正交试验,确定最佳合成工艺条件。
1.3.3 高效液相色谱分析
色谱柱:Waters SugarPak I;流动相:超纯水;流速0.4 mL/min;柱温85 ℃;进样量10 μL。
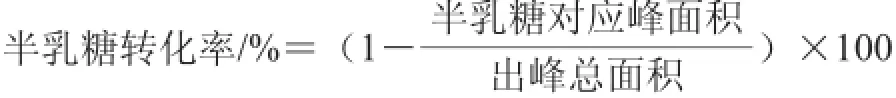
1.3.4 分离、纯化
将所得半乳聚糖粗产品用去离子水溶解,然后在溶解液中缓慢加入5 倍去离子水体积的无水乙醇(99%),并不断搅拌,离心,弃上清液,得半乳聚糖沉淀。然后将沉淀用少量去离子水溶解后冷冻干燥,得不含半乳糖、催化剂及引发剂的半乳聚糖。
乙醇沉淀后的半乳聚糖用去离子水溶解,然后用Sephadex G-25葡聚糖凝胶色谱柱分离,以去离子水作洗脱剂,流速0.5 mL/min,用部分收集器收集洗脱液,每管收集量为3 mL,时间间隔6 min。收集液中的糖组分采用硫酸-苯酚法测定[13],检测波长为490 nm,以吸光度为纵坐标,绘制洗脱曲线。收集各管洗脱液,旋转蒸发浓缩后,冻干得白色粉末状样品。
1.3.5 纯度和分子质量
参照吕志华等[14]报道的方法进行测定。
1.3.6 单糖组成分析
纯化后的半乳聚糖用三氟乙酸水解,方法参照Wang Hui等[15]的报道。水解液去除三氟乙酸后,采用高效阴离子交换色谱(high performance anion exchange chromatography,HPAEC)法分析单糖组成。方法参照朱松等[16]的报道。
1.3.7 红外光谱测定
参照张汇等[17]采用的方法进行测定。
1.3.8 氢核磁共振波谱(1H-nuclear magnetic resonance,1H-NMR)测定
参照刘小如等[18]采用的方法进行测定。
2 结果与分析
2.1 反应温度、微波辐照时间、引发剂浓度、催化剂添加量对半乳糖转化率的影响
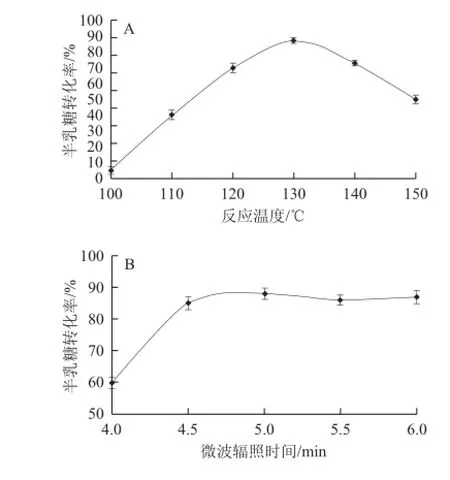
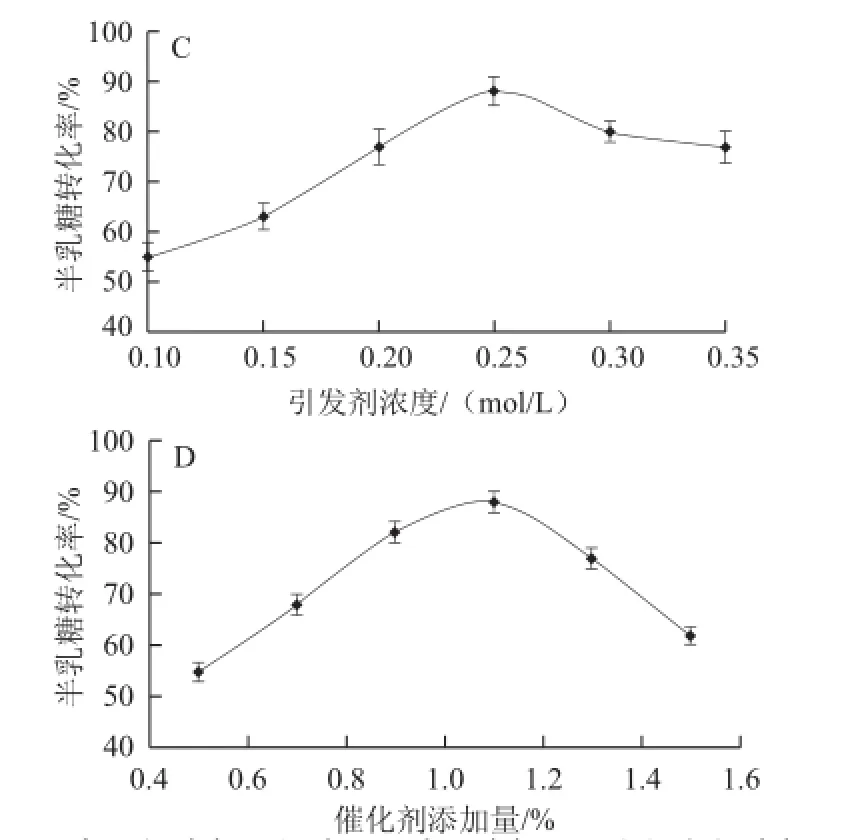
图1 反应温度(A)、微波辐照时间(B)、引发剂浓度(C)以及催化剂添加量(D)对半乳糖转化率的影响Fig.1 Effects of reaction temperature (A), microwave irradiation time (B), initiator concentration (C) and catalyst doage (D) on galactose conversion rate
糖在酸的催化作用下加热可脱水缩合生成聚糖[19]。温度在此缩合反应中起着重要作用,本实验采用微波提供能量,由于微波具有一定的穿透能力,并可直接对物料进行加热,相比于传统的间接加热方式,微波辐照可使物料受热均匀,从而产生均一的化学反应。图1A为引发剂浓度0.3 mol/L、催化剂添加量1.1%、微波辐照5 min条件下温度变化对半乳聚糖合成的影响,由图1A可见,反应温度为130 ℃时,半乳糖转化率可达到91%。
反应时间是半乳聚糖制备过程中的一个重要影响因素,本实验考察反应温度130 ℃,引发剂浓度0.3 mol/L,催化剂添加量1.1%时,时间变化对半乳聚糖合成的影响,由图1B可见,半乳糖的转化率随反应时间的延长而提高,当反应时间为4.5 min时,转化率达到最高,随着反应时间的延长,半乳糖的转化率保持平稳,可能是反应后期,随着体系中水分的蒸发,酸浓度提高,产物半乳聚糖的酸水解反应和原料半乳糖的酸催化缩合反应达到平衡。
微波发出能量的快速、有效吸收,对于加速、提高半乳糖的缩合反应速率至关重要。在本实验的反应体系中,由于底物半乳糖为固态,不能吸收微波能量,杂多酸催化剂虽为液态并且具有极性,但为微量添加,不能很好的吸收微波能量,启动缩合反应,因此,缩合反应需要能够有效吸收微波能量的引发剂。氯化钠溶液作为引发剂使用具有快速吸收微波能量,迅速提高反应体系温度,以及不影响缩合反应进行等优点。本实验研究了引发剂氯化钠溶液的浓度变化对半乳糖转化率的影响。结果表明,在反应温度130 ℃、催化剂添加量1.1%、反应时间5 min、引发剂浓度0.3 mol/L时,缩合反应转化率达到最大值,如图1C所示。随引发剂浓度的增加,半乳糖转化率降低,原因可能是一定反应时间下,高浓度引发剂快速吸收能量,使反应达到半乳糖缩合与半乳聚糖水解的平衡点,但随着反应时间的延长,半乳聚糖的水解占优势,因此半乳糖的转化率降低。
杂多酸作为催化剂来加速半乳糖的缩合反应,首先在酸催化作用下半乳糖质子化成为糖基供体,未质子化的半乳糖作为糖基供体,二者在特定反应温度条件下脱水缩合形成半乳糖聚合物。中间产物水作为极性溶剂,可辅助引发剂吸收微波能量,但由于反应温度(130 ℃)高于水的沸点,因此水分蒸发。当催化剂添加量增加时,在特定反应时间(5 min)条件下,酸水解半乳聚糖反应速率高于酸催化半乳糖的缩合反应,因此当催化剂添加量高于1.1%时,半乳糖的转化率降低,如图1D所示。
2.2 正交试验结果

表1 正交试验设计及结果Table 1 Orthogonal experimental design and results
由表1可知,极差R的大小为A>D>C>B,A对试验结果影响最大,B影响最小。通过正交试验得最佳合成条件为A3B2C4D3,在反应温度130 ℃、微波辐照4.5 min、引发剂浓度0.25 mol/L、催化剂添加量1.1%时,半乳糖转化率最高为97.22%。
2.3 合成产物高效液相色谱分析

图2 合成产物高效液相色谱图Fig.2 HPLC of synthetic products
如图2所示,微波辅助合成半乳聚糖产物中,半乳聚糖约占92.23%,低聚半乳糖约占4.99%,半乳糖占2.78%(保留时间13.724 min)。半乳糖转化率为97.22%。
2.4 合成产物的分离、纯化
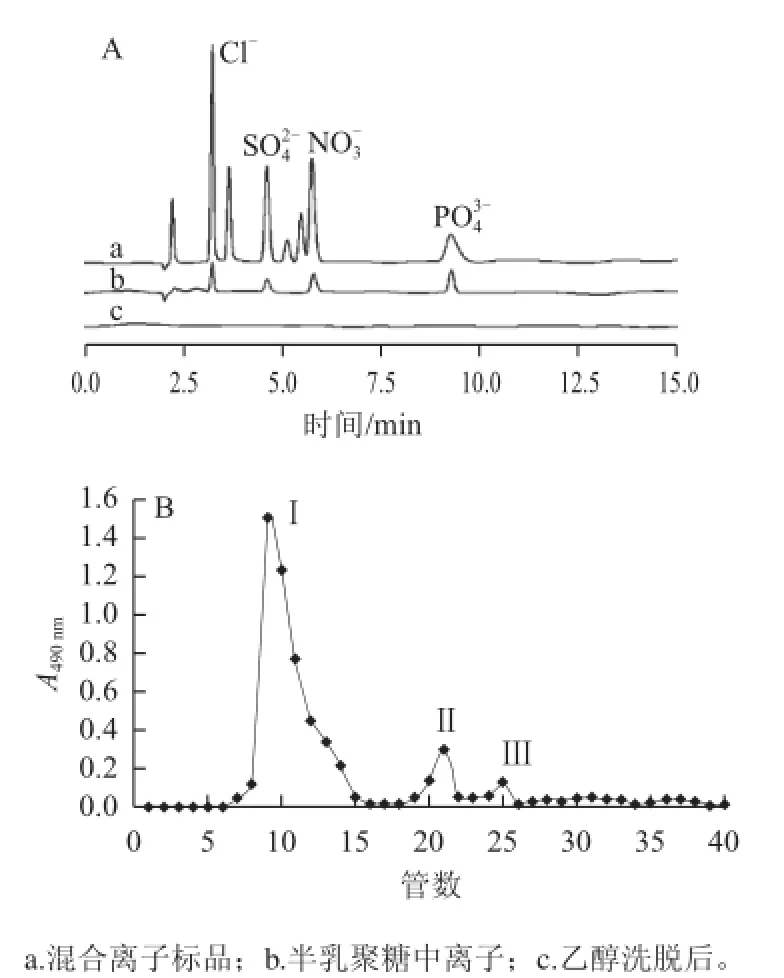
图3 合成产物乙醇沉淀后的HPAEC图(A)以及乙醇沉淀后半乳聚糖的Sephadex G-25洗脱曲线(B)Fig.3 HPAEC of synthetic products after ethanol precipitation (A) and Sephadex G-25 elution curve of galactan precipitated by ethanol (B)
微波辅助合成的半乳聚糖产物中含有催化剂及引发剂,乙醇洗脱可以将其有效去除。如图3A所示,经乙醇洗脱后,半乳聚糖产物中的杂多酸得到有效去除。乙醇洗脱后浓缩干燥的半乳糖经Sephadex G-25葡聚糖凝胶柱分析,如图3B所示,得3 个洗脱峰,将峰Ⅰ对应洗脱液收集并浓缩干燥,得半乳聚糖葡聚糖凝胶柱分离样品。
2.5 分子质量测定
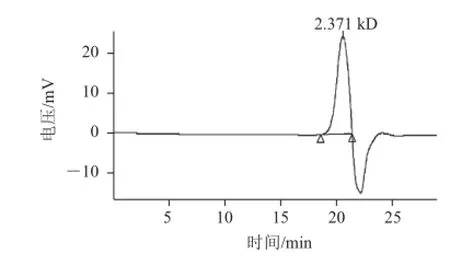
图4 半乳聚糖的高效凝胶渗透色谱图Fig.4 HPGPC of galactan
高效凝胶过滤色谱分析葡聚糖凝胶柱分离的半乳聚糖样品,得到单一、狭窄、对称的色谱峰,表明收集到的半乳聚糖为均一组分。如图4所示,半乳聚糖的重均分子质量(Mw)为2.853 kD,数均分子质量(Mn)为2.347 kD,峰位分子质量(Mp)为2.371 kD,分子质量分布系数(HI=Mw/Mn)为1.21,平均聚合度为17。HI表明分离纯化后的半乳聚糖分子质量的多分散程度小,微波辅助合成的主要产物(半乳聚糖)分子质量分布均一。
2.6 单糖组成分析
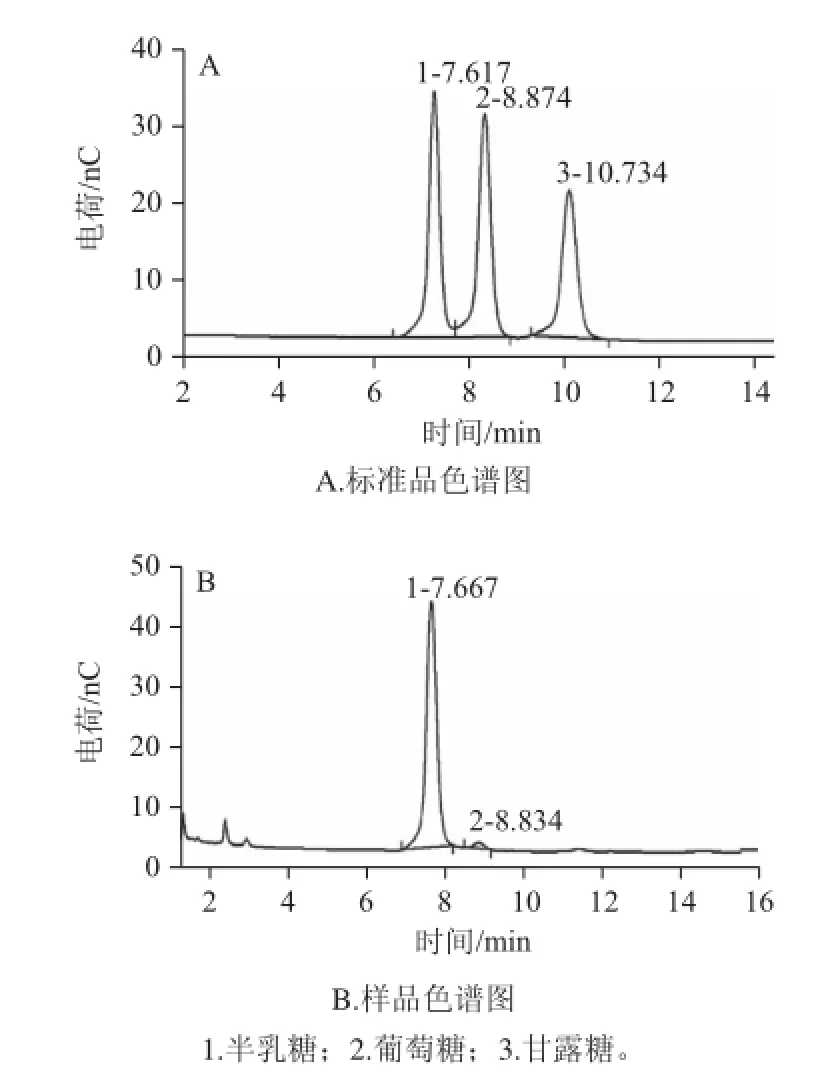
图5 半乳聚糖三氟乙酸水解物的HPAECC分析Fig.5 HPAEC analysis of galactan hydrolyzed by TFA
半乳聚糖经三氟乙酸水解,HPAEC分析表明,如图5所示,半乳聚糖水解物由半乳糖(保留时间7.667 min)和微量葡萄糖(保留时间8.834 min)组成。此结果表明半乳糖在杂多酸催化下微波辐照脱水缩合生成了半乳聚糖。三氟乙酸水解后产生微量葡萄糖的原因是由于酸的作用使少量半乳糖分子异构化为葡萄糖,因此HPAEC图谱上显示有微量葡萄糖峰。
2.7 红外光谱分析

图6 半乳聚糖的红外光谱图Fig.6 IR spectrum of galactan
由图6可见,3 000~3 700 cm-1处有强且宽的吸收峰,是由O—H的伸缩振动产生。3 406 cm-1和2930 cm-1处的吸收峰分别由羟基(—OH)和C—H的伸缩振动产生[20], 1638、1572 cm-1处为—C=O的吸收峰,1 411 cm-1处的吸收峰为—C—O振动产生[21]。1 144、1 081、1 036 cm-1处为半乳聚糖的特征吸收峰,875 cm-1和797 cm-1处的吸收峰表明半乳聚糖中存在α-和β-构型,695 cm-1处的小峰为半乳糖的特征吸收峰。
2.81H-NMR光谱分析
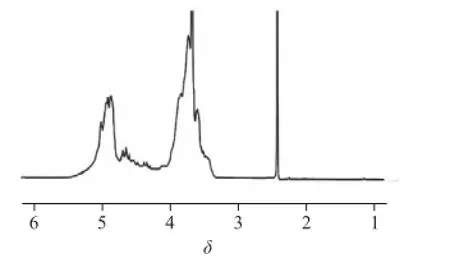
图7 半乳聚糖的1H-NMR(400 MHz、255 ℃)Fig.7 400 MHz1H-NMR spectrum of galactan recorded at 25 ℃
1H-NMR能够提供糖的α-或β-异头碳的构型信息,对于两种构型的端基质子,大多数的α-端基质子化学位移出现在δ 5~6之间,而大多数β-端基质子化学位移出现在δ 4~5之间,H-2到H-6信号出现在δ 3.2~4.5之间[22]。如图7所示,半乳聚糖绝大多数端基质子的信号出现在δ 4~5之间,少部分端基质子信号出现在δ 5~6之间,表明半乳聚糖以β-构型为主,同时有少部分的α-构型,与红外光谱分析结果相符。
3 结 论
采用微波辅助酸催化的方法能够快速、高效的合成半乳聚糖。合成条件为引发剂浓度0.25 mol/L、催化剂添加量1.1%、反应温度130 ℃、微波辐照4.5 min,半乳糖转化率为97.22%。合成产物经乙醇沉淀脱离子,及葡聚糖凝胶色谱柱分离得到平均分子质量为2.853 kD,平均聚合度为17的聚半乳糖,离子交换色谱分析聚半乳糖的单糖组成为半乳糖及微量葡萄糖。红外光谱及1H-NMR分析表明,半乳聚糖具有多糖的结构特征,并且以β-构型为主。本研究可为多糖的快速合成及其衍生物的制备提供新的研究思路。
[1] DELATTRE C, FENORADOSOA T A, MICHAUD P. Galactans: an overview of their most important sourcing and applications as natural polysaccharides[J]. Brazilian Archives of Biology and Technology, 2011, 54(6): 1075-1092.
[2] GRIESHOP C M, FLICKINGER E A, FAHEY G C Jr.,. Oral administration of arabinogalactan affects immune status and fecal microbial populations in dogs[J]. Nutrition, 2002, 132(3): 478-482.
[3] CARLUCCI M J, SCOLARO L A, ERREA M I, et al. Antiviral activity of natural sul phated galactans on herpes virus multiplication in cell culture[J]. Planta Medicine, 1997, 63(5): 429-432.
[4] CHATTOPADHYAY K, MATEU C G, MANDAL P, et al. Galactan sulfate of Grateloupia indica: isolation, structural features and antiviral activity[J]. Phytochemistry, 2007, 68(10): 1428-1435.
[5] FARIAS W R L, VALWNT A P, PEREIRA M S, et al. Structure and anticoagulant activity of sulphated galactans: isolation of a unique sulphated galactan from the red algae Botryocladia occidentalis and comparison of its anticoagulant action with that of sulphated galactans from invertebrates[J]. Journal of Bological Chemistry, 2000, 275(38): 299-307.
[6] COOMBE D R, PARISH C R, RAMSHAW I A, et al. Analysis of the inhibition of tumour metastasis by sulphated polysaccharides[J]. International Journal of Cancer, 1987, 39(1): 82-88.
[7] FALSHAW R, BIXLER H J, JOHNDRO K. Structure and performance of commercial-2 carrageenan extracts. Part Ⅲ. Structure analysis and performance in two dairy applications of extracts from the New Zealand red seaweed, Gigartina atropurpurea[J]. Food Hydrocolloids, 2003, 17(2): 129-139 .
[8] 唐仕荣, 刘全德, 苗敬芝, 等. 两种微波辅助萃取法萃取牛蒡多糖[J].食品科学, 2009, 30(18): 102-105.
[9] 曹崇江, 鞠兴荣, 刘晓庚. 微波协同固体超强酸合成肉桂酸异丙酯[J].食品科学, 2013, 34(24): 1-5.
[10] 王海松, 乐国伟, 丁苏, 等. 微波固相合成葡-半乳低聚糖的工艺研究[J].食品工业科技, 2008, 29(5): 182-184.
[11] GEORGE C. Chemistry of foods and beverages: recent developments[M]//ALLINGHAM R P. Polydextrose-a new food ingredient: technical aspects. Elsevier Inc., 1982: 293-303.
[12] BAKER C W. Production of sucrose-based carbohydrates for the food industry[J]. Food Technology, 1993, 47: 149-150.
[13] 郭金龙, 陈有君, 孙国琴, 等. 苯酚-硫酸法测定杏鲍菇多糖方法的研究[J]. 食品科学, 2008, 29(12): 555-558.
[14] 吕志华, 于广利, 赵峡, 等. 不同标准品对HPGPC法测定多糖相对分子质量的影响[J]. 中国新药杂志, 2002, 11(3): 220-221.
[15] WANG Hui, LIU Gang, ZHOU Benhong, et al. Monosaccharide compositional analysis of purified polysaccharide from Tricholoma matsutake by capillary gas chromatography[J]. Journal of Medicinal Plants Research, 2012, 6(10): 1935-1940.
[16] 朱松, 戴军, 陈尚卫, 等. 高效阴离子交换色谱法检测酱油中的单糖及双糖[J]. 分析测试学报, 2012, 31(11): 1411-1415.
[17] 张汇, 王君巧, 聂少平, 等. 黑灵芝葡聚糖高活性硫酸化产物的制备及分离纯化[J]. 食品科学, 2013, 34(23): 128-132.
[18] 刘小如, 张美丽, 胡蒋宁, 等. 油茶粕多糖的分级纯化及结构研究[J].食品科学, 2013, 34(23): 96-102.
[19] MANLEY-HARRIS M, RICHARDS G N. A novel fructoglucan from the thermal polymerization of sucrose[J]. Carbohydrate Research, 1993, 240(40): 183-196.
[20] LIU Chunhui, LIN Qinxiong, GAO Yi, et al. Characterization and antitumor activity of a polysaccharide from Strongylocentrotus nudus eggs[J]. Carbohydrate Polymers, 2006, 67(3): 313-318.
[21] F ENG Hao, LI Jian, WANG Lijuan. Preparation of biodegradable flax shive cellulose-ba sed superabsorbent polymer under microwave irradiation[J]. BioResources, 2010, 5(3): 1484-1495.
[22] CUI S W. Food carbohydrates chemistry, physical properties, and applications[M]. CRC Press: Taylor & Fra ncis Group, 2005: 154-155.
Microwave-Assisted Synthesis and Structural Analysis of Galactan
WANG Hai-song1, SHI Yong-hui1,2, LE Guo-wei1,2,*
(1. Research Center of Food Nutrition and Functional Factors, School of Food Science and Technology, Jiangnan University, Wuxi 214122, China; 2. State Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi 214122, China)
Purpose: To develop a rapid and effi cient method to synthesize galactan and analyze the structure of the synthetic product. Methods: For preparation of galactan, gal actose was allowed to undergo a dehydration-condensation reaction initiated by sodium chloride solution and catalyzed by heteropolyacid under m icrowave irradiation. The synthetic product was preci pitated by addition of anhydrous ethanol and purified/fractionated by Sepherdex G-25 column chromatography. The purity and degree of polymerization (DP) were analyzed by high performance gel permeation chromatography (HPGPC), the monosaccharide composition by high performance anion exchange chromatography (HPAEC), and the structure by IR and NMR spectroscopy. Results: The optimal conditions for synthesizing galactan were determined as follows: initiator concentration 0.25 mol/L, catalyst dosage 1.1%, reaction temperature 130 ℃ and microwave irradiation time 4.5 min. Under these conditions, the conversion rate of galactose was 97.22%, and the synthesized product had an average molecular weight of 2.853 kD and a degree of polymerization of 17 and was composed of monosaccharide components such as galactose and trace glucose. IR and1H-NMR analysis showed that the sugar residues in the galactan were dominated by β-confi guration. Conclusion: Galactan can be rapidly and effi ciently synthesized by microwave irradiation under the catalysis of heteropolyacid.
microwave; galactan; structural analyses
TS202.3
A
1002-6630(2014)24-0035-05
10.7506/spkx1002-6630-201424007
2014-03-17
“十二五”国家科技支撑计划项目(2012BAD33B05)
王海松(1981—),男,博士研究生,研究方向为食品营养与功能因子。E-mail:hswang201166@gmail.com
*通信作者:乐国伟(1956—),男,教授,博士,研究方向为食品营养与功能因子。E-mail:lgw@jiangnan.edu.cn
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
天然产物研究与开发(2019年10期)2019-11-05
中成药(2018年8期)2018-08-29
中成药(2018年3期)2018-05-07
中国调味品(2017年2期)2017-03-20
中国民族医药杂志(2016年2期)2016-05-14
中华老年多器官疾病杂志(2016年7期)2016-04-28
中学化学(2015年2期)2015-06-05
国际心血管病杂志(2015年5期)2015-02-27
化工生产与技术(2014年3期)2014-02-27
