
能量代谢信号通路对缺血性脑卒中神经保护作用的研究进展
2014-03-08 02:07:45王云杰侯卓然廖红张陆勇庞涛
药学进展 2014年9期
王云杰,侯卓然,廖红,张陆勇,庞涛
(1.中国药科大学江苏省新药筛选重点实验室,江苏 南京 210009;2. 江苏大学医学院,江苏 镇江 212013)
能量代谢信号通路对缺血性脑卒中神经保护作用的研究进展
王云杰1,侯卓然2,廖红1,张陆勇1,庞涛1*
(1.中国药科大学江苏省新药筛选重点实验室,江苏 南京 210009;2. 江苏大学医学院,江苏 镇江 212013)
据近几年研究发现,能量代谢通路中的关键酶,如AMPK、Sirt1、PGC-1α、NAMPT,在控制缺血再灌注引起的钙离子超载、兴奋性氨基酸毒性、过氧化应激等损伤途径中发挥重要的作用,并且对缺血性神经起到保护作用。分别介绍这些能量代谢通路中的关键蛋白在缺血性神经保护中的作用机制,旨在为缺血性脑卒中的治疗提供新思路。
能量代谢通路;缺血性脑卒中;神经保护
缺血性脑卒中发病率居高不下,其高致死率和高致残率给人类健康和生活带来巨大影响,也对社会造成了沉重的经济负担[1]。缺血再灌注(IRI)损伤作为一种严重而复杂的病理过程,涉及到离子失衡、过氧化应激、神经兴奋性毒性及炎症激活等负性事件的参与,这些级联瀑布效应最终导致神经细胞的坏死和凋亡,致使不可逆性神经功能缺失。目前医学界唯一的有效治疗脑缺血的方法是溶栓疗法,然而,使用溶栓药物的时间窗限制了其在临床中的应用[2]。因此,寻找有效可靠的神经保护剂成为治疗脑卒中的当务之急。研究发现,缺血性脑卒中对大脑缺血性神经的损伤主要是因为能量供应不足,从而引起星形胶质细胞的糖酵解增加来提供能量,但长期糖酵解过程造成乳酸堆积增加,并且抑制神经元利用乳酸作为能量来源,使神经细胞死亡[3]。单磷酸腺苷(adenosinemonophosphate,AMP)激活的蛋白激酶(AMPK)作为能量代谢通路中的关键酶,控制着整个机体的能量代谢;沉默信息调节因子2相关酶1(silentmatingtypeinformationregulator2homolog1,Sirt1)是一种尼克酰胺腺嘌呤二核苷酸(nicotinamideadeninedinucleotide,NAD)依赖的Ⅲ型组蛋白去乙酰基酶,通过调节NAD的含量对各种细胞因子及AMPK进行脱乙酰化作用,进而减轻缺血再灌注引起的损伤;过氧化物酶体增殖活性受体(peroxisomeproliferatorreceptor,PPAR)γ辅助因子1α(PPARγcoactivator1α,PGC-1α)是过氧化物酶体增殖物激活受体PPAR的转录共同激活因子,主要通过增加线粒体的合成以及其他的转录因子的活性,对缺血性神经起保护作用;尼克酰胺磷酸核糖转移酶(nicotinamidephosphoribosyltransferase,NAMPT)作为NAD补救合成通路中的限速酶,不仅通过NAD的合成保护神经细胞,并且可能作为一种细胞因子,影响炎症发生过程。因此,如果从这些能量代谢通路上的关键蛋白入手,调控能量代谢过程,将对缺血性神经产生重要的保护作用。
1 AMPK对缺血性神经的保护作用
AMPK广泛存在于真核细胞生物中,属于丝氨酸/苏氨酸蛋白激酶。AMPK在能量状态发生改变的条件下(如ATP含量的下降、AMP/ATP比例的升高)被激活,来调节很大范围内的细胞活动并重建新陈代谢的平衡。当AMPK被激活时,快速地使下游靶标磷酸化,使ATP消耗通路阻断,且促进ATP合成通路的开放,因此,AMPK被称为“细胞能量调控器”[4]。以下对AMPK在缺血性脑卒中发生时的所发挥的作用进行说明。
1.1 在大脑缺血时对缺血性神经的保护作用
很多新陈代谢的变化,创伤或毒性作用对神经系统造成伤害后都能够引起AMPK的激活,包括大脑缺血、低血糖症、受代谢毒素的影响等情况,此时AMPK的α1、α2、β1、β2和γ1亚基在神经细胞中高度表达[5]。
AMPK激活剂5-氨基咪唑-4-甲酰胺核苷酸转甲酰酶(AICAR)在一些模型中显示,对大脑缺血时的神经元具有保护作用。这表明AMPK激活可以促使神经细胞能量的恢复。有实验证实:加入低浓度的AICAR时,AMPK被激活,并且对抗缺糖损伤、新陈代谢变化、兴奋性毒性和氧化应激引起的神经元损伤,从而起到保护神经细胞的作用。更重要的是,在低浓度AICAR处理的原代细胞中,AICAR的这种保护作用被证明与AMPK的短暂激活有关[6]。Moss及其领导的科研团队发现,AMPK与γ-氨基丁酸(GABAB)受体有直接的联系,且使胞质尾区的S783磷酸化,这个过程可能会刺激GABA依赖的机制,使突触前Ca2+释放受阻[7],从而抑制Ca2+超载的发生,减轻缺血再灌注引起的缺血损伤。
然而,也有关于AMPK激活在神经保护和抗细胞凋亡方面作用的不同意见。一些研究表明,AMPK的延长激活会有不利影响,并且激活细胞坏死机制。体内外实验表明,AMPK对神经细胞作用的差异可能是由葡糖糖浓度的不同引起。因此,应用相同的新陈代谢条件,已有实验室证明了AMPK的激活对暴露在代谢应激、钙超载、兴奋性中毒情况下的神经细胞有保护作用。
1.2 在其他方面对缺血性神经的保护
首先,AMPK对身体的代谢有调节作用。它能抑制脂肪酸和胆固醇的合成,促进糖酵解,抑制糖异生,为神经细胞提供ATP。并且,AMPK是HMG-CoA还原酶的上游调节因子,能很好地改善模型动物和临床脑卒中病人的神经损伤[8]。其次,AMPK通过抑制神经元型一氧化氮合酶(nNOS),减少了过硝酸盐(ONOO-)的产生,发挥神经保护作用[9]。此外,AMPK通过促进血管内皮生长因子(VEGF)的合成,促进内皮细胞的增殖和代谢[10]。最后,AMPK参与神经细胞的自噬活动,介导巨噬细胞M1型和M2型的转化,清除细胞内受损伤的细胞结构、衰老的细胞器以及不再需要的生物大分子,为细胞内各种生理活动提供能量[11]。
AMPK对缺血性神经的保护机制如图1所示。

图1 AMPK对缺血性神经的保护机制Figure 1 Protection mechanism of AMPK on ischemic nerve
2 Sirt1对缺血性神经的保护作用
Sirt1是一种NAD依赖的Ⅲ型组蛋白去乙酰基酶,广泛存在于各种细胞及器官中,例如细胞核、胞浆和线粒体[12]。它的酶活力取决于它的蛋白表达水平、NAD的含量和调节酶活性的蛋白的水平,例如,当饥饿或细胞暴露于氧化应激和DNA损伤的环境中时,它的表达量会升高。最近的研究发现:实验性脑缺血后,梗死区域的Sirt1含量明显增加;Sirt1的活性影响梗死面积的大小,Sirt1基因敲除使脑卒中的后果加重[13]。基于以上实验发现,人们对Sirt1在脑缺血引起的神经损伤中的作用机制展开了进一步研究。接下来,从以下3个方面就Sirt1对缺血性神经的保护机制进行分析。
2.1 Sirt1在缺血时对缺血性神经的保护
首先,Sirt1通过激活AMPK产生作用。AMPK系统激活,作为代谢和应激信号转导的原件,调节下游的靶蛋白,发挥生物学效应。缺血时的低能量状态导致了AMPK的激活,且有研究发现葡萄糖含量变化引起的AMPK的变化与NAD+/NAD的比例以及Sirt1的水平和活力有关[14]。
其次,另一个触发细胞保护且防止缺氧引起代谢改变的因素是缺氧诱导因子(HIF)。在氧不足时,HIF2α的水平显著上调,并且它的活性受Sirt1脱乙酰化作用的影响。另外,Sirt1与HIF1α相互作用,Sirt1抑制HIF1α的转录活性。在低氧应激时,胞内NAD的减少抑制Sirt1对刺激的反应,增加HIF1α乙酰化作用,并因此增加HIF1α基因的表达。有趣的是,其他研究发现HIF2α与HIF1α竞争与Sirt1的作用[15]。
最后,在代谢控制中与Sirt1最密切的一个因子是PGC-1α。Sirt1与PGC1α在功能上相互作用并且使PGC-1α脱乙酰化,从而引起线粒体蛋白包括ATP合成通路的过量表达。PGC-1α通过消除活性氧(ROS)和诱导解耦联蛋白2(UCP2)等起到神经保护作用[16]。
Sirt1对缺血损伤的保护机制如图2所示。

图2 Sirt1对缺血损伤的保护机制Figure 2 Protective mechanism of Sirt1 on ischemic injury
2.2 Sirt1在再灌注时对缺血性神经的保护
缺血对神经损伤较大,但更严重的损伤是再灌注造成的。在再灌注过程中,细胞的新陈代谢回到有氧途径,产生ROS。ROS主要产生于线粒体并触发多种机制,包括Ca2+的积累,半胱天冬酶激活,细胞因子上调,以及脂质、蛋白质和DNA损伤。ROS生成和消除失衡产生氧化应激[17]。
心肌细胞的各种研究报告显示了Sirt1对抗氧化应激的保护作用。心肌过度表达Sirt1对缺血再灌注(IRI)时的抗氧化应激作用有很强的对抗性,是因为Sirt1上调抗氧化剂如超氧化物歧化酶(MnSOD)和硫氧还蛋白-1的表达起作用的。Sirt1也使Forkhead转录因子FoxO1脱去乙酰基,诱导其核易位和随后的抗氧化分子的转录[18]。此外,在肾脏IRI模型中,Sirt1对抗氧化应激的保护作用也被证实。NO是由内皮NO合成酶(eNOS)合成,是内皮功能的关键调节器,它对抗内皮素的血管收缩作用从而引起血管舒张。然而,由iNOS产生的NO加剧了IRI损伤时过氧化物或过氧硝酸盐的生成。有大量的证据支持Sirt1和eNOS之间的关系,Sirt1交互并调节eNOS的乙酰化状态,导致酶的激活[19]。
Sirt1对再灌注损伤的保护机制如图3所示。

图3 Sirt1对再灌注损伤的保护机制Figure 3 Protective mechanism of Sirt1 on reperfusion injury
2.3 Sirt1对炎症的抑制作用和抗细胞凋亡作用
IRI导致组织炎症反应,是由于免疫细胞浸润组织引起的。各种细胞因子、趋化因子、黏附分子和细胞外基质化合物可以终止炎症的损害。这些因素的表达是由特定的核转录因子κB(NF-κB)介导的,它是一个关键的炎症调节器。激活后,转录因子向核迁移,并且促进促炎基因转录,加强炎症反应[20]。Sirt1对炎症的抑制是通过抑制NF-κB,并激活内皮eNOS改善微循环,从而抑制炎症的发生[21]。
Sirt1抑制细胞凋亡是通过多个途径实现的,例如,抑制p53的转录活性或增加Ku70和Bax之间的绑定作用[22]。
Sirt1在抑制炎症和抗细胞凋亡中的作用如图4所示。
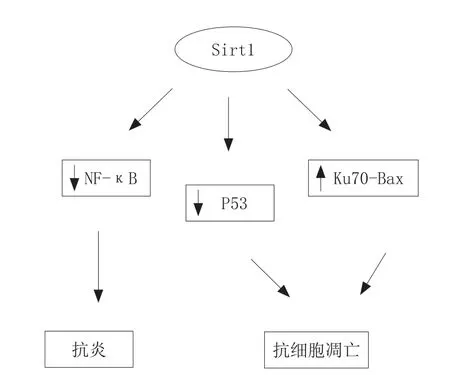
图4 Sirt1在抑制炎症和抗细胞凋亡中的作用Figure 4 The role of Sirt1 in inhibiting inflammation and apoptosis
3 PGC-1α对缺血性神经的保护作用
PGC-1α是PPAR的转录共激活因子[23]。PGC-1α具有明显的组织特异性,主要在富含线粒体或者能量要求高的组织如大脑、骨骼肌中表达,其他组织中表达很少[24]。PGC-1α对转录因子有强大的控制性,最近,有研究发现PGC-1α具有神经保护作用,PGC-1α敲除小鼠缺血后脑神经元的损伤显著,增加PGC-1α的表达,使得培养的细胞免受氧化应激介导的细胞死亡。以下对PGC-1α在缺血性神经保护方面的可能机制进行概述。
3.1 PGC-1α促进线粒体的合成
在过表达PGC-1α的神经细胞中,线粒体的含量增加。PGC-1α调节核呼吸因子-1(NRF-1)和核呼吸因子-2(NRF-2),使得线粒体转录因子A表达,调节线粒体的溶解和破裂。此外,PGC-1α还可以通过诱导解偶联蛋白2(UCP-2)的产生,促进线粒体的生物合成。而且,PGC-1α本身还可以增强线粒体的呼吸功能,使得缺血性神经元的能量供给大大增加,使神经细胞在缺血时存活下来的机会增加[25]。
3.2 PGC-1α消除氧自由基
PGC-1α可通过上调UCP-2和超氧化物歧化酶2(SOD2)的水平,减少ROS的释放。因为,在缺血动物模型中,上调UCP2,活性氧的释放和神经元的损伤明显减少,说明UCP2减少ROS的释放,对神经元起保护作用。并且,过表达SOD2对局灶性脑缺血后的氧化应激诱导的神经损伤也具有保护作用[26]。
3.3 PGC-1α的抗细胞凋亡作用
半胱天冬酶(Caspase)和Bcl-2蛋白对细胞凋亡起到调控作用。近来,在体外培养的神经细胞中进行的研究表明,PPAR-γ激动剂罗格列酮可减少氧糖缺乏模型(OGD)中细胞的凋亡数目,减少细胞色素C的释放,维持线粒体膜电位稳定,抑制蛋白激酶9的活性。而且,使用罗格列酮或过表达PPAR-γ可提高SOD的酶活力,减少ROS的生成,最终增加抗凋亡家族Bcl-2和Bcl-xL的含量,减少蛋白激酶B的降解,从而阻止细胞凋亡[27]。
3.4 PGC-1α抑制炎症反应
PGC-1α具有较强的抗炎作用,可能是通过干扰激活蛋白-1(AP-1)、NF-κB、转录激活因子(STAT)的反式激活能力,进而调节细胞因子产生和黏附分子的表达来减轻炎症[28]。
3.5 PGC-1α促进血管生成
PGC-1α促使缺血组织的血管生成,可能是通过分泌大量血管生长因子(包括VEGF等)起作用[29]。
图5描述了PGC-1α对缺血性神经的保护机制。

图5 PGC-1α对缺血性神经的保护机制Figure 5 Protection mechanism of PGC-1α on ischemic nerve
4 NAMPT对缺血性神经的保护作用
NAMPT是NAD补救合成途径中的限速酶[30],也曾被称为B细胞克隆增强因子(PBEF)和内脏脂肪素(visfatin),是哺乳动物NAD合成的主要途径,起调控细胞内NAD水平的作用[31]。NAMPT广泛存在于细胞核、细胞浆及线粒体中,这些统称为细胞内NAMPT(intracellularNAMPT,iNAMPT),NAMPT还可以通过旁分泌和内分泌的方式释放到细胞外,称为细胞外NAMPT(extracellularNampt,eNAMPT),eNAMPT主要是血清中的NAMPT[32]。
以下就NAMPT对缺血性神经保护作用的最新报道进行介绍。
4.1 胞内NAMPT对缺血性神经的保护作用
随着年龄增加,NAD的水平在不同组织(包括大脑)中均会持续下降。NAMPT在脑内主要表达于神经元细胞,而在神经胶质细胞不表达[32]。实验显示:1)在大脑中动脉堵塞(MCAO)模型和氧糖剥夺(OGD)模型中,NAMPT的水平显著上调,而NAMPT特异性抑制剂FK866可加重MCAO模型动物的脑损伤和缺氧缺血导致的神经元损伤,但这些现象可以被NAMPT的终产物烟酰胺单核苷酸(NMN)逆转[33];2)AMPK的激活有神经保护作用,过表达的NAMPT可以进一步增加缺血或氧糖剥夺所致的AMPK的激活,而干扰RNA可以抑制NAMPT,从而抑制AMPK的激活[32];3)NAMPT可以通过NAD的合成量来调控Sirt1的去乙酰化能力,AMPK的上游激酶LKB1,它的乙酰化和细胞内的重分布均受Sirt1的调节[34];4)MCAO术后,NAMPT过表达的大鼠脑内自噬小泡的数目增加,mTOR和S6K1的磷酸化水平下降,mTOR-S6K1信号通路系由TSC2调节,研究发现TSC2的Ser1387位点磷酸化水平增强,而NAMPT敲除大鼠表现出自噬能力下降,自噬阻断剂阻断NAMPT过表达引起的神经保护作用,而诱导剂加强其作用。因此,NAMPT可通过改变TSC2-Ser1387-mTOR-S6K1信号通路,在缺血早期增加自噬的反应[35]。
综上所述,iNAMPT对缺血性神经的保护作用通过2个途径实现:1)NAMPT可通过其酶活性,控制NAD的合成,影响Sirt1的活性,调节LKB1的去乙酰化和细胞内重分布,最终调控AMPK的激活,减少神经元在缺血情况下的凋亡和死亡;2)NAMPT可通过改变TSC2-Ser1387-mTOR-S6K1信号通路,在脑缺血早期增加自噬的发生,以提高神经元在缺血情况下的存活率。
iNAMPT对缺血性神经的保护机制如图6所示。
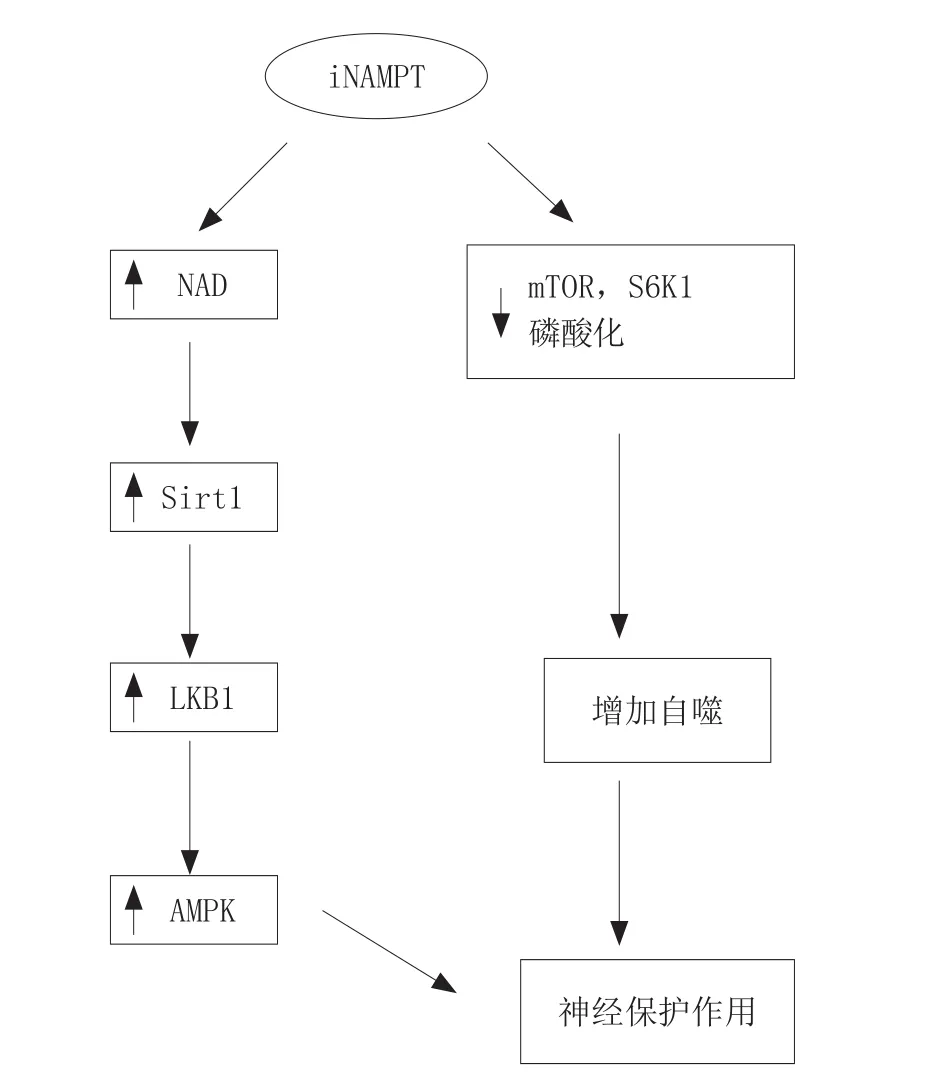
图6 iNAMPT对缺血性神经的保护机制Figure 6 Protection mechanism of iNAMPT on ischemic nerve
4.2 胞外NAMPT对缺血性神经的保护作用
相比于iNAMPT,eNAMPT对缺血性神经作用的研究较晚,并且eNAMPT没有典型的分泌信号[36],它可以通过一些细胞的非典型分泌机制分泌到细胞间隙或血液中,这些细胞包括脂肪细胞、肝细胞、巨噬细胞和白血球细胞[37]。有研究指出OGD时,神经元NAMPT过表达,通过初级神经元释放到胞外介质中;分泌的NAMPT保护少突胶质细胞(OL)免于OGD的损伤,而OL的主要功能是在中枢神经系统中包绕轴突、形成绝缘的髓鞘结构、协助神经电信号的跳跃式高效传递,以及维持和保护神经元的正常功能[38]。
eNAMPT对神经的保护作用与NAD依赖的保护作用不同,它既可能是通过促进胞外NAD的生物合成来发挥作用,也可能是通过一种未知的功能,像是绑定在一种特殊的受体上,并因此激活信号通路。与此一致,有文献报道,随着NAMPT重组蛋白的表达,可以激活一些促存活通路,包括PBK/Akt、ERK1/2和STAT5a,并且这些分子可能具有调节NAMPT促神经保护的作用。换言之,eNAMPT可能绑定于一个细胞膜上的未知受体,通过激活下游的促活信号通路发挥它的神经保护作用[39]。
除了作为NAD的一个限速酶的作用外,eNAMPT也能以细胞因子的角色来抑制外周系统的炎症。NAMPT基因最初是从外周血液cDNA文库中分离出来的,NAMPT表现出对干细胞因子以及白介素-7的前B细胞因子生成的激活作用[40]。其可控制单核细胞和中性粒细胞在炎症刺激中的作用,炎症的作用在大脑缺血模型中被完善,且在大脑损伤和修复的过程中起重要作用[41]。同样地,炎症的急性过度应激过程可能对神经细胞有损害作用,然而,一个减弱的延长的炎症效应,对神经修复发挥有利的作用,这是至关重要的。因此,Zheng等[39]推测eNAMPT不仅影响OL,也与缺血性脑卒中后脑内小胶质细胞(中枢神经系统的炎症细胞)的炎症发生过程有关,并且对缺血性脑卒中发生后脑内炎症的发生有抑制作用,但实验结果未证实这一推测,还需进一步研究。最近有研究显示eNAMPT可以通过能量限制(CR)对抗氧化应激,发挥其抗氧化应激的作用[42]。
综上所述,eNAMPT对神经的保护作用通过2个方面实现:1)促进胞外NAD的合成;2)作为细胞因子,影响脑内小胶质细胞的炎症发生过程。当然,对eNAMPT在缺血性神经保护方面的作用,还需展开进一步研究。
图7描述了eNAMPT对缺血性神经的保护机制。
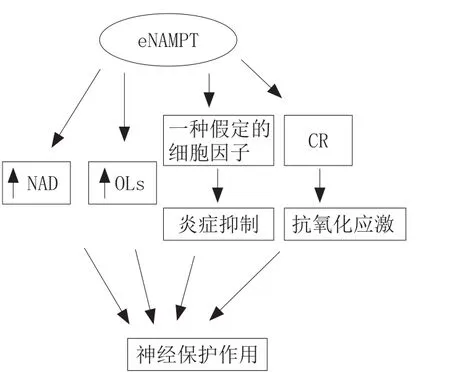
图7 eNAMPT对缺血性神经的保护机制Figure 7 Protection mechanism of eNAMPT on ischemic nerve
5 结语
越来越多的研究者认为:治疗缺血性脑卒中,需从神经本身出发,寻找修复和重建损伤神经的新方法,而不仅仅是应对缺血引起的损伤。能量代谢中的关键蛋白在最近的研究中表现出对缺血性神经重要的保护作用,虽然它们的机制还不甚明确,需要进一步探索,但是我们相信,能量代谢通路中关键蛋白调节剂的研究与开发,可以为缺血性脑卒中的治疗提供一个新的选择,其不仅能够避免溶栓治疗引起的出血危害,还可以对缺血发生时引发的损伤进行及时的阻断以及对缺血再灌注后引起的神经损害进行有效的补救。可以期待,随着对能量代谢通路中这些关键蛋白调节机制不断深入的研究,缺血性脑卒中新的治疗方法必将造福于更多的患者。
[1]Juan P, Bolanos, Maria A, et al. Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: therapeutic implications [J]. Adv Drug Deliv Rev, 2009, 61 (14): 1299-1315.
[2]Deb P, Sharma S, Hassan K M. Pathophysiologic mechanisms of acute ischemicstroke:anoverviewwithemphasisontherapeuticsignifcancebeyond thrombolysis [J]. Pathophysiology, 2010, 17 (3): 197-218.
[3]Blázquez C1, Woods A, de Ceballos M L, et al. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes [J]. J Neurochem, 1999, 73 (4): 1674-1682.
[4]Hardie D G, Scott J W, Pan D A, et al. Management of cellular energy by the AMP-activated protein kinase system [J]. FEBS Lett, 2003, 546 (1): 113-120.
[5]Turnley A M, Stapleton D, Mann R J, et al. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system [J]. J Neurochem, 1999, 72 (4): 1707-1716. [6]Weisová P, Dávila D, Tuffy L P, et al. Role of 5'-adenosine monophosphateactivated protein kinase in cell survival and death responses in neurons [J]. Antioxid Redox Signal, 2011, 14 (10): 1863-1876.
[7]Kuramoto N, Wilkins M E, Fairfax B P, et al. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase [J]. Neuron, 2007, 53 (2): 233-247.
[8]Sillesen H, Amarenco P, Hennerici M G, et al. Atorvastain reduces the risk of cardiovasular envents in patients with carotid atherosclerosis: a secondary analysis of the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) trial [J]. Stroke, 2008, 39 (12): 3297-3302.
[9]Zou M H, Hou X Y, Shi C M, et al. Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase [J]. J Biol Chem, 2002, 277 (36): 32552-32557.
[10]Zou M H, Mu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function [J]. Clin Exp Pharmacol Physiol, 2008, 35 (5/6): 535-545.
[11]Wang P, Guan Y F, Du H, et al. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia [J]. Autophagy, 2012, 8 (1): 77-87.
[12]Denu J M. The Sir 2 family of protein deacetylases [J]. Curr Opin Chem Biol, 2005, 9 (5): 431-440.
[13]Nakagawa T, Guarente L. Sirtuins at a glance [J]. J Cell Sci, 2011, 124 (Pt 6): 833-838.
[14]Ruderman N B, Xu X J, Nelson L, et al. AMPK and SIRT1: a longstanding partnership? [J]. Am J Physiol Endocrinol Metab, 2010, 298 (4): E751-E760.
[15]Lim J H, Lee Y M, Chun Y S, et al. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha [J]. Mol Cell, 2010, 38 (6): 864-878.
[16]St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators [J]. Cell, 2006, 127 (2): 397-408.
[17]Raedschelders K, Ansley D M, Chen D D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion [J]. Pharmacol Ther, 2012, 133 (2): 230-255.
[18]Vinciguerra M, Santini M P, Martinez C, et al. mIGF-1/JNK1/SirT1 signaling confers protection against oxidative stress in the heart [J]. Aging Cell, 2012, 11 (1): 139-149.
[19]Schmitt C A, Heiss E H, Dirsch V M. Effect of resveratrol on endothelial cell function: molecular mechanisms [J]. Biofactors, 2010, 36 (5): 342-349. [20]Stein S, Matter C M. Protective roles of SIRT1 in atherosclerosis [J]. Cell Cycle, 2011, 10 (4): 640-647.
[21]Petegnief V, Planas A M. SIRT1 regulation modulates stroke outcome [J]. Transl Stroke Res, 2013, 4 (6): 663-671.
[22]Hernández-Jiménez M, Hurtado O, Cuartero M I, et al. Silent information regulator 1 protects the brain against cerebral ischemic damage [J]. Stroke, 2013, 44 (8): 2333-2337.
[23]BentonCR,WrightDC,BonenA.PGC-1α-mediatedregulationofgene expression and metabolism: implications for nutrition and exercise prescriptions [J]. Appl Physiol Nutr Metab, 2008, 33 (5): 843-862.
[24]Tritos N A, Mastaitis J W, Kokkotou E G, et al. Characterization of the peroxisomeproliferatoractivatedreceptor1alpha(PGC-1α)expressionin the murine brain [J]. Brain Res, 2003, 961 (2): 255-260.
[25]Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha [J]. Cardiovasc Res, 2008, 79 (2): 208-217.
[26]Raha S, McEachern G E, Myint A T, et al. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase [J]. Free Radic Biol Med, 2000, 29 (2): 170-180.
[27]Chen S D, Yang D I, Lin T K, et al. Roles of oxidative stress, apoptosis, PGC-1αandmitochondrialbiogenesisincerebralischemia[J].Int J Mol Sci, 2011, 12 (10): 7199-7215.
[28]Delerive P, Fruchart J C, Staels B, et al. Peroxisome proliferatoractivatedreceptorsininfammationcontrol[J]. J Endocrinol, 2001, 169 (3): 453-459.
[29]Arany Z, Foo S Y, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha [J]. Nature, 2008, 451 (7181): 1008-1012.
[30]Sethi J K, Vidal-Puig A. Visfatin: the missing link between intra-abdominal obesity and diabetes? [J]. Trends Mol Med, 2005, 11 (8): 344-347.
[31]Samal B, Sun Y, Stearns G, et al. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor [J]. Mol Cell Biol, 1994, 14 (2): 1431-1437.
[32]Wang P, Vanhoutte P M, Miao C Y. Visfatin and cardio-cerebro-vascular disease [J]. J Cardiovasc Pharmacol, 2012, 59 (1): 1-9.
[33]Adya R, Tan B K, Chen J, et al. Pre-B cell colony enhancing factor (PBEF)/ visfatin induces secretion of MCP-1 in human endothelial cells: role in visfatin-induced angiogenesis [J]. Atherosclerosis, 205 (1): 113-119.
[34]Fulco M, Cen Y, Zhao P, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt [J]. Dev Cell, 2008, 14 (5): 661-673.
[35]Wang P, Guan Y F, Du H, et al. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia [J]. Autophagy, 2012, 8 (1): 77-87.
[36]Friebe D, Neef M, Kratzsch J, et al. Leucocytes are a major source of circulating nicotinamide phosphoribosyltransferase (NAMPT)/pre-B cell colony(PBEF)/visfatinlinkingobesityandinfammationinhumans[J].Diabetologia, 2011, 54 (5): 1200-1211.
[37]Garten A, Petzold S, Barnikol-Oettler A, et al. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes [J]. Biochem Biophys Res Commun, 2010, 391 (1): 376-381.
[38]McDonald J W, Althomsons S P, Hyrc K L, et al. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity [J]. Nat Med, 1998, 4 (3): 291-297.
[39]Jing Z, Xing J, Chen X, et al. Neuronal NAMPT is released after cerebral ischemia and protects against white matter injury [J]. J Cereb Blood Flow Metab, 2014, 34 (10): 1613-1621.
[40]Schubert K, Gutknecht D, Köberle M, et al. Melanoma cells use Thy-1 (CD90) on endothelial cells for metastasis formation [J]. Am J Pathol, 2013, 182 (1): 266-276.
[41]Gelderblom M, Leypoldt F, Steinbach K, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke [J]. Stroke, 2009, 40 (5): 1849-1857.
[42]Song J, Ke S F, Zhou C C, et al. Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation [J]. J Gerontol A Biol Sci Med Sci, 2014, 69 (1): 44-57.
Advances in Research on Neuroprotective Role of Energy Metabolism Signaling Pathway in Ischemic Stroke
WANG Yunjie1, HOU Zhuoran2, LIAO Hong1, ZHANG Luyong1, PANG Tao1
( 1. Jiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing 210009, China; 2.School of Medicine, Jiangsu University, Zhenjiang 212013, China)
Recent researches have shown that the key enzymes of energy metabolism pathways, such as AMPK、Sirt1、PGC-1α、NAMPT, can play key roles in resisting calcium overload, excitatory amino acid toxicity and peroxide stress during ischemia reperfusion injury, and protect ischemic nerve. The neuroprotection mechanisms of the key enzymes of energy metabolism pathways have been introduced in this paper, aiming at providing a new method for the treatment of ischemic stroke.
energy metabolism pathway; ischemic stroke; neuroprotection
R966
A
1001-5094(2014)09-0665-07
接受日期:2014-10-20
项目资助:国家自然科学基金项目(No.21402241),江苏省自然科学基金项目(No.BK20130653);
* 通讯作者:庞涛,副研究员;
研究方向:药物筛选及新药开发;
Tel: 025-83271043; E-mail: tpang@cpu.edu.cn
猜你喜欢
自然杂志(2021年6期)2021-12-23 08:24:46
中成药(2021年5期)2021-07-21 08:39:04
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:05:56
现代装饰(2018年5期)2018-05-26 09:09:01
西南军医(2016年6期)2016-01-23 02:21:19
电源技术(2015年5期)2015-08-22 11:18:38
弹箭与制导学报(2015年1期)2015-03-11 15:32:06
发明与创新(2015年37期)2015-02-27 10:40:25
西南军医(2015年2期)2015-01-22 09:09:37
癌变·畸变·突变(2014年3期)2014-03-01 04:39:48
