
金红石型TiO2(110)表面甲醇氧化机制的理论模拟
2014-03-02 02:15郎秀峰
河北科技师范学院学报 2014年4期
郎秀峰
(河北科技师范学院物理系,河北秦皇岛,066004)
TiO2通常作为一种催化剂或光催化剂,特别是在有机分子的光降解和光分解水领域,被广泛地使用和研究[1]。简单醇类也常被用作模型体系来研究TiO2表面有机污染物的热氧化和光氧化过程[1]。在这些醇类物质中,因为CH3OH可以作为一种空穴吸收剂来极大地提高TiO2光催化分解水的能力,所以不同条件下,CH3OH在TiO2表面的光化学行为受到了人们的广泛关注[1~3]。许多关于CH3OH在TiO2表面选择性氧化的工作已经发现了不同的产物,如H2,CO2,H2CO,HCOOH,HCOOCH3[4]。
金红石型TiO2(110)表面是一个典型的模型[5],在这个表面上对于CH3OH的热化学行为已经开展了许多实验和理论研究。温控脱附光谱和扫面隧道显微镜都证实了在TiO2(110)表面CH3OH的分子形式和分解形式都是可以存在的,且它分解为CH3O的过程一般都是在桥位的氧空位(VO)和表面的原子台阶上进行的[1,6,7]。但是对于CH3OH在TiO2(110)表面的五配位钛原子(Ti5c)上是否能够进行热分解目前还存在很大的争论。近年来,一些试验也开始关注CH3OH在Ti5c上的光化学行为[8~10]。这些试验通常是通过光照TiO2(110)表面吸附的CH3OH分子,然后使用温控脱附光谱和扫描隧道显微镜来观测光反应的产物。试验上已经发现了 CH3OH分子在光照条件下可以产生 CH3O,H2CO和HCOOCH3。关于CH3OH分子在TiO2(110)表面的氧化机制,试验和理论已经证实CH3OH分子首先经过羟基脱氢生成了CH3O,然后经过甲基基团的脱氢进一步转化成H2CO[11],但是对于H2CO如何进一步生成HCOOCH3的过程了解得还不是很多。例如,Friend等[9]提出H2CO在光照条件下首先生成了甲酰基,然后HCO与CH3O结合生成了HCOOCH3。但是,Yang等[10]通过CH3OH的同位素试验研究建议H2CO直接和CH3O结合生成CH3OC(=O)H2,进一步脱氢生成了HCOOCH3。尽管存在一些争论,但是关于TiO2(110)表面的甲醇氧化过程的理论研究迄今为止还几乎没有开展过。
基于上述研究,通过第一性原理计算,系统地探索了完美和有缺陷的TiO2(110)表面上CH3OH氧化为HCOOCH3的反应机制。通过调查CH3OH氧化过程中所有基元反应的动力学,证实了基元反应的动力学能够很清楚地识别出一条反应路径,即在完美的TiO2(110)表面上,H2CO选择性地生成了CH3OC(=O)H2而不是HCO,并最终转化成了HCOOCH3。
1 理论方法
本研究中的电子结构计算是在密度泛函理论(Density Functional Theory,DFT)的框架下完成的,主要使用了VASP软件[12]。其中,交换和相关能是采用一般梯度近似(Generalized gradient approximation,GGA)下的PBE泛函来完成的[13]。笔者分别为Ti原子、C原子和O原子选取了12,4和6个价电子,这些价电子与核的作用是通过投影放大波(Projector augmented-wave,PAW)[14]方法来处理的。所有表面反应都是在(4×2)的TiO2(110)表面平板体系中完成的。表面平板模型包括了12个原子层和15 Å的真空层。在结构优化过程中,模型中表面吸附物质和顶部的9层原子是被允许弛豫的,而底部的3层原子距离是被固定在TiO2体相材料中原子之间的距离。使用3.85×104kJ/mol的截断能,计算过程中体系总能量标准设定为0.96×10-3kJ/mol。基于每个基元反应的初始和终态物质的优化结构,通过裸露弹性带方法(Climbing image nudged elastic band,CI-NEB)[15]计算了最小能量路径和相应的活化能。
2 结果和讨论
比较CH3OH通过中间物HCO和CH3OC(=O)H2生成HCOOCH3的2种机制。在这2种机制中,CH3OH 氧化的主要产物为 CH3O,H2CO,HCO,CH3OC(=O)H2和 HCOOCH3。
2.1 甲醇及其氧化物在TiO2(110)的吸附
TiO2(110)-(4×2)表面上甲醇及其氧化产物 CH3O,H2CO,HCO,CH3OC(=O)H2和 HCOOCH3的稳定几何构型(图1)。这些物质相对应的Ti5c-O键的键长列在表1中。从这些优化构型中可以发现CH3OH中的氧原子吸附于Ti5c原子上,与氧原子相连的氢原子指向了表面上最临近的桥位氧原子(Obr)。CH3O的吸附构型与CH3OH的吸附构型非常相近。它们主要的差别是在CH3O吸附构型中,原来CH3OH分子中与O相连的H转移到表面的Obr上,并且CH3基团有点向表面的法线方向倾斜。在H2CO的吸附构型中,CH2基团对角地指向了表面[001]方向的桥位氧。在HCO分子中,分子平面垂直于桥位氧列,并且它的H和O分别于邻近的 Obr原子和 H形成了2个氢键。CH3OC(=O)H2和HCOOCH3采用了相似的构型,其中分子的骨架平行于桥位氧列。以前的理论研究已经报道了CH3OH,CH3O和H2CO在TiO2(110)表面的吸附构型[11,16],与本次研究得到的吸附构型是完全一致的。但是,对于HCO,CH3OC(=O)H2和HCOOCH3在TiO2(110)表面的吸附构型至今还没有进行过理论模拟研究。
为了比较包含这些物质的基元反应从热力学角度进行的难易度,通过下面的公式计算了这些物质在 TiO2(110)表面的吸附能[16]:
其中,ETiO2和ECH3OH是纯TiO2(110)表面和自由的CH3OH分子的能量,E吸附分子/TiO2是CH3OH及其氧化产物吸附于TiO2(110)表面的吸附结构的总能量。这里物质吸附能的值越小代表这种物质越稳定。表1列出了各种物质的吸附能。从表中数据可以发现,2个CH3OH分子的吸附能是-76.18 kJ/mol。当1个CH3OH分子失去氢转变为CH3O时,混合的CH3OH和CH3O构型的吸附能变为-73.29 kJ/mol,仅仅比CH3OH分子的吸附能高了2.89 kJ/mol。当2个 CH3OH分子都失去氢原子转变为CH3O后,体系的吸附能变为-56.90 kJ/mol,这比CH3OH分子的吸附能高了19.28 kJ/mol。比较相近的吸附能说明CH3OH可以以分子态和分解态的形式出现在TiO2(110)表面。过去的研究也报道了CH3OH和CH3O在TiO2(110)的吸附能是很接近的[16]。但是这些研究中2个CH3OH或CH3O是相互作用的,计算得到的吸附能包含这些分子之间的作用。与过去的研究相比,本计算选择了更大的表面模型,避免了分子之间的相互作用,同时选择了更高的计算精度。这些都保证了计算得到的是完全独立的CH3OH分子的吸附能。
当CH3O进一步失去H转变为H2CO后,体系的吸附能变为-5.78 kJ/mol,比CH3O的吸附能高出了51.11 kJ/mol。说明这种反应从能量角度来说是很难进行的。对于含有中间产物HCO的反应路径,H2CO转变为CH3O使得吸附能从-5.78 kJ/mol变为83.90 kJ/mol。这表明了从H2CO转变为CH3O在热力学上是极难进行的。因为从HCO转变为HCOOCH3时吸附能降低为7.71 kJ/mol,所以这个反应是可以自发进行的。对于含有CH3OC(=O)H2的反应路径,CH3OC(=O)H2较低的吸附能(-27.00 kJ/mol)表明H2CO和CH3O直接耦合生成CH3OC(=O)H2是可以自发进行的,但是CH3OC(=O)H2进一步脱H转变为HCOOCH3在热力学上是很难进行的。

图1 TiO2(110)-(4×2)表面上CH3OH及其氧化产物构型的俯视和侧视图
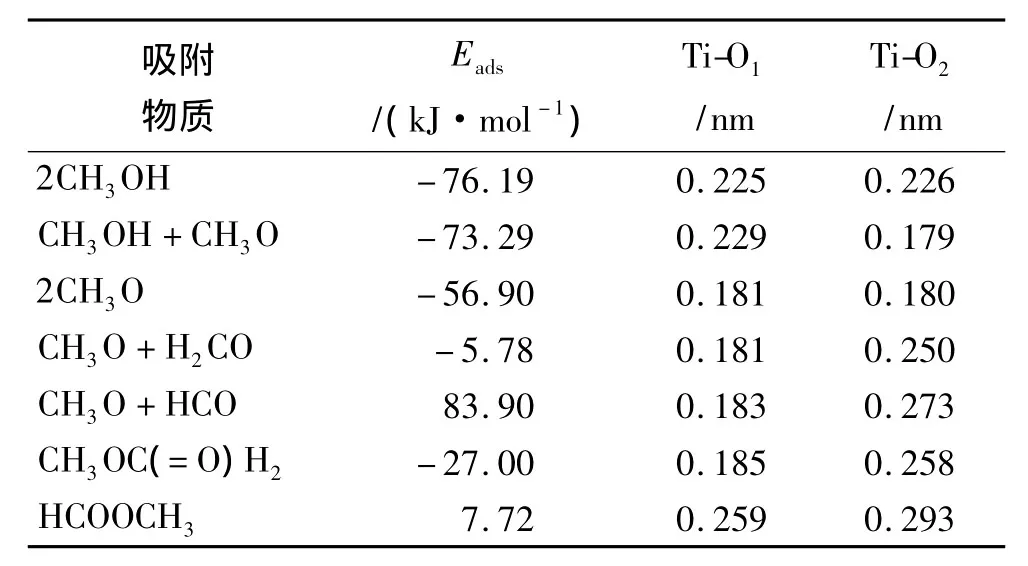
表1 CH3OH及其氧化产物在TiO2(110)表面的吸附能(Eads)和Ti5c-O键的键长
2.2 CH3OH在TiO2(110)表面的氧化路径
在确定了CH3OH及其氧化产物在TiO2(110)表面的稳定吸附构型后,进一步探索了CH3OH在TiO2表面转变为HCOOCH3的反应能剖面。对于初始的2个反应步,CH3OH分子通过一个O-H键的断裂分解成CH3O,而CH3O进一步通过1个C-H键的断裂转变为H2CO。2个反应可以通过下面2个化学反应式描述:

2个CH3OH分子进行上述反应的势能剖面如图2(a)所示。图中每个构型的能量都是相对于TiO2(110)表面吸附2个CH3OH分子的总能来计算的。计算所得的各个反应的能垒和热力学能列于表2中。第1个CH3OH分子的O-H分解步拥有1个33.75 kJ/mol的能垒。在这步反应中,过渡态1有1个强的Obf-H键(0.117 nm)和一个弱的O-H键(0.129 nm),它的构型非常接近产物CH3O。由于CH3OH转变为CH3O的能垒比较小,所以这个反应应该是很容易进行的。对于第2个CH3OH分子的分解来说,虽然过渡态2构型与过渡态1构型非常相似,但是第2个CH3OH分子分解为CH3O需要1个相对大的能垒(46.29 kJ/mol)。在2个CH3OH都转变为CH3O之后,其中1个CH3O通过将1个H转移给表面的邻近的Obr而进一步分解为甲醛。甲氧基的C-H键的断裂需要跨越一个很大的能垒(169.73 kJ/mol)(图2)。对比计算得到的O-H键和C-H键断裂的反应能垒可以得出一个结论:在周围环境中对CH3OH进行热激发就可以让其转变为CH3O,而CH3O须在光激发的条件下才可以开始转变为H2CO。Henderson等运用温控脱附谱图证实了CH3OH可以通过热反应生成CH3O,而CH3O需要经过光照才能转变成H2CO[8]。另外,杨学明等通过第一性计算证实了CH3OH反应生成甲氧基的势垒远低于CH3O转化为H2CO的势垒[11]。这些试验和理论报道与本研究得到的结果是完全一致的。生成的H2CO在TiO2(110)表面吸附比较弱,可以很容易地扩散到与CH3O相邻的位置,这个过程可以从图2(a)的过渡态4的低势垒(24.11 kJ/mol)得到证实。
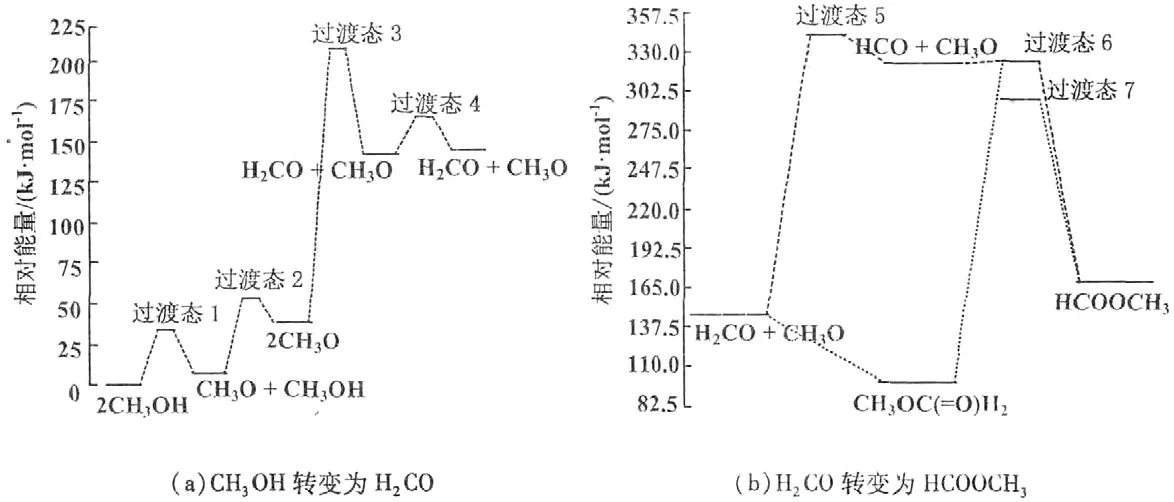
图2 TiO2(110)表面CH3OH转变为HCOOCH3的势垒剖面
2个相邻的H2CO和CH3O可以经过2种反应路径产生HCOOCH3。这2种反应路径可以用下面4个化学反应式来表述:

反应式中(4)和(5)代表了包含中间物HCO的反应路径I,反应式(6)和(7)代表了包含中间物CH3OC(=O)H2的反应路径II。这些基元反应的势能剖面显示在图2(b)中,且相关的能垒和热力学数据能列于表2中。
在包含中间物HCO的反应路径I中,H2CO的C-H键的断裂需跨越一个大的能垒(196.73 kJ/mol)才可以发生。相应的过渡态5的原子构型包含了一个强的Obr-H键(0.097 5 nm),且产生的HCO基团正倾向于与邻近的表面Obr原子形成1个氢键。HCO在形成之后,它的羰基基团中缺电子的C会去攻击CH3O的富电子的O,进而形成最终的产物HCOOCH3。与上述基元反应为吸热过程不同,这个反应经历了一个放热过程,只需要跨越一个很小的势垒(1.93 kJ/mol)就可以发生,说明从 HCO到HCOOCH3在热力学和动力学上都是可行的。
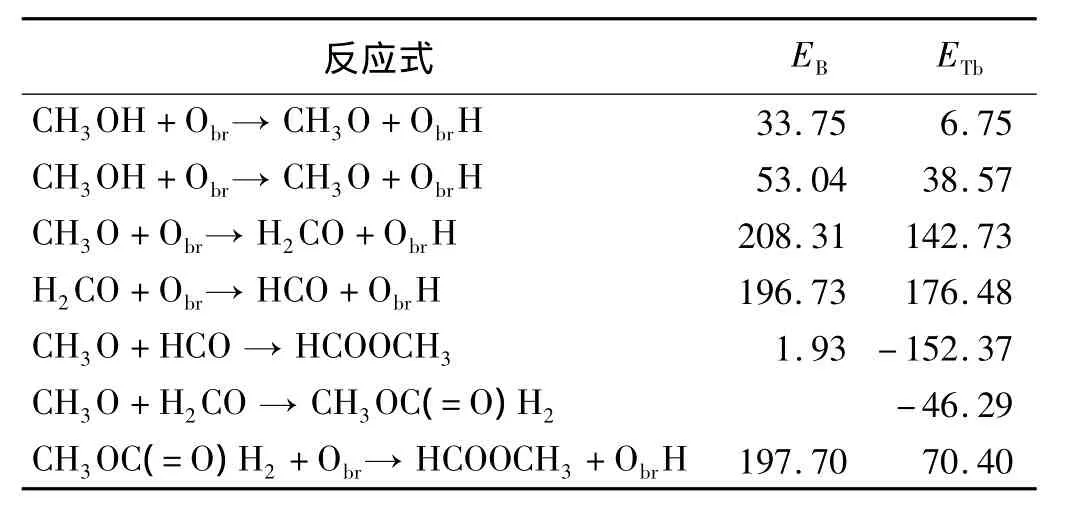
表2 TiO2(110)表面上CH3OH氧化所经历的可能反应路径的能垒EB和热力学能ETb kJ·mol-1
在包含中间物CH3OC(=O)H2的反应路径II中,H2CO和CH3O的直接耦合也包含了H2CO中羰基上的缺电子C对CH3O中富电子O的攻击过程。这个反应过程也是放热过程,不需要跨越能垒就可以完成。这说明CH3OC(=O)H2的产生是可以以很高的反应速率自发进行的。但是,CH3OC(=O)H2通过CH2O基团中1个C-H键断裂生成HCOOCH3的过程需要很大的能量来跨越一个大的能垒(196.74 kJ/mol)。过渡态7中氢的转移过程通过一个短的Obr-H键键长(0.113 nm)和一个长 C-H 键键长(0.162 nm)表现。总体来说,H2CO分子或者CH3OC(=O)H2分子的C-H键断裂在CH3OH转变为HCOOCH3的整个氧化过程中具有相对大的能垒,表明这种C-H键断裂的反应在这2种反应途径中是决定整个氧化过程速率的基元反应。
因为反应路径I和II中决速反应具有非常相近的能垒,所以从动力学角度分析,从CH3OH到HCOOCH3的氧化过程2种反应路径都是可行的。这2种反应路径的关键选择还需要通过H2CO对HCO或CH3OC(=O)H2的转化率来确定。CH3OC(=O)H2的产生过程是放热的,不需要跨越能垒(表2)。相比之下,HCO的产生过程需要吸收大量的热,且需要跨过很大的势垒。这些差异表明从热力学和动力学角度来说,H2CO更倾向于生成CH3OC(=O)H2而不是HCO,然后再进一步生成HCOOCH3。
对于CH3OH转化为H2CO的反应机制,过去已经进行了很多研究,人们对它的认识比较深。相比之下,对于H2CO转化为HCOOCH3的理论和试验研究比较少,人们对于它们的反应机制还非常不了解。如上所述,最近,2个实验小组同时观察到吸附于TiO2(110)表面的CH3OH在光照条件下最终生成HCOOCH3[9,11],但是他们对H2CO转化为HCOOCH3的反应机制提出了不同的观点。笔者首次通过计算证实了CH3OH在TiO2(110)表面主要是通过H2CO和CH3O的直接耦合来生成HCOOCH3,很好地解释了试验上存在的争论。
3 结 论
基于第一性原理计算详细地研究了TiO2(110)表面CH3OH氧化生成HCOOCH3的反应机制。对这个反应过程中可能经历的包含中间物HCO和CH3OC(=O)H2等2种反应路径的所有基元反应进行了动力学分析。计算所得的各个基元反应的能垒清楚地证明了2种反应路径在动力学上都是可行的,且H2CO更倾向于生成CH3OC(=O)H2而不是HCO。这些基元反应的动力学行为证实了CH3OH在TiO2(110)表面的氧化主要是通过H2CO与CH3O耦合生成CH3OC(=O)H2,然后进一步脱氢产生HCOOCH3。这些结果可能帮助人们更深刻地认识TiO2表面CH3OH的完整的氧化机制,并可能提供一条绿色的、对环境友好的由醇类和醛类直接转化为脂类的合成路径。
[1] Henderson M A.A surface science perspective on TiO2photocatalysis[J].Surf Sci Rep,2011,66(6-7):185-297.
[2] Kominarni H,Sugahar H,Hashimoto K.Photocatalytic selective oxidation of methanol to methyl formate in gas phase over titanium(IV)oxide in a flow-type reactor[J].Catal Commun,2010,11(5):426-429.
[3] Panayotov D A,Burrows S P,Morris J R.Photooxidation mechanism of methanol on rutile TiO2nanoparticles[J].J Phys Chem C,2012,116(11):6 623-6 635.
[4] Chiarello G L,Ferri D,Selli E.Effect of the CH3OH/H2O ratio on the mechanism of the gas-phase photocatalytic reforming of methanol on noble metal-modified TiO2[J].J Catal,2011,280(2):168-177.
[5] Henderson M A,Lyubinetsky I.Molecular-Level insights into photocatalysis from scanning probe microscopy studies on TiO2(110)[J].Chem Rev,2013,113(6):4 428-4 455.
[6] Zhang Z,Bondarchuk O,White J M,et al.Imaging adsorbate O-H bond cleavage:methanol on TiO2(110)[J].J Am Chem Soc,2006,128(13):4 198-4 199.
[7] Bates S P,Gillan M J,Kresse G.Adsorption of Methanol on TiO2(110):a first-principles investigation[J].J Phys Chem B,1998,102(11):2 017-2 026.
[8] Shen M,Henderson M A.Identification of the Active Species in Photochemical Hole Scavenging Reactions of Methanol on TiO2[J].J Phys Chem Lett,2011,2(21):2 707-2 710.
[9] Phillips K R,Jensen S C,Baron M,et al.Sequential photo-oxidation of methanol to methyl formate on TiO2(110)[J].J Am Chem Soc,2013,135(2):574-577.
[10] Guo Q,Xu C,Yang W,et al.Methyl formate production on TiO2(110),initiated by methanol photocatalysis at 400 nm[J].J Phys Chem C,2013,117(10):5 293-5 300.
[11] Guo Q,Xu C,Ren Z,et al.Stepwise photocatalytic dissociation of methanol and water on TiO2(110)[J].J Am Chem Soc,2012,134(32):13 366-13 373.
[12] Kresse G,Gurthmüller J.Efficient iterative schemes for ab initio-energy calculations using a plane-wave basis set[J].Phys Rev B,1996,54:11 169-11 186.
[13] Perdew J P,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Phys Rev Lett,1996,77:3 865-3 868.
[14] Kresse G,Joubert D.From ultrasoft Pseudopotentials to the projector augmented-wave method[J].Phys Rev B,1999,59:1 758-1 775.
[15] Henkelman G,Jónsson H.Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points[J].J Chem Phys,2000,113:9 978-9 985.
[16] Zhao J,Yang J,Petek H.Theoretical study of the molecular and electronic structure of methanol on a TiO2(110)surface[J].Phys Rev B,2009,80:235 416.
[17] Xu B,Haubrich J,Baker T A,et al.Theoretical study of O-assisted selective coupling of methanol on Au(111)[J].J Phys Chem C,2011,115:3 703-3 708.
(责任编辑:石瑞珍)
猜你喜欢
计算机工程与应用(2023年1期)2023-01-13
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
科学导报(2018年30期)2018-05-14
北京航空航天大学学报(2017年10期)2017-04-20
航天返回与遥感(2014年4期)2014-07-31
无机化学学报(2014年4期)2014-02-28
