
弥漫性掌跖角化症一家系11例报告
2014-02-21 13:43:30杨云东钟庆坤
云南医药 2014年5期
纳 猛,李 霞,杨云东,钟庆坤,宋 杰,张 娴,王 婵
(开远市人民医院 皮肤科,云南 开远 661699)
掌跖角化症(palmoplantar keratoderma,PPK)是一组以掌跖部呈弥漫性或局限性角化过度的先天性遗传病,包括十几种类型,可见于人类各种族或民族,弥漫性掌跖角化症(dif-fuse epidemolytic palmopantar keratodema diffuse EPPK) 又称局部表皮松解性角化过度症(localized epidemolytic hy-perkeratosis,OMM# 144200),是一种最常见的掌跖角化症亚型。其主要特征是出生后或幼儿期发病,持续终生的手部、足跖部皮肤弥漫性浅黄色角质增厚层。本文收集了弥漫性掌跖角化症一家系11例患者,报道如下。

图1、2

图3、4
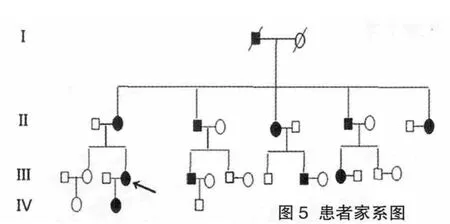
图5 患者家系图
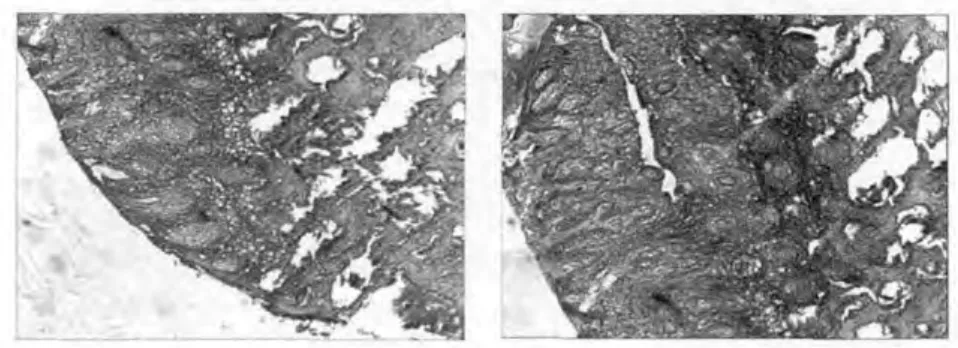
图6、7
病例与家系 先证者Ⅲ-4,普某,女,22岁,农村居民,籍贯云南省蒙自市。因双手掌、双足跖皮肤长期增厚、皲裂、多汗,经多年治疗无效;而其2岁女儿于出生后6月开始双手掌、足跖皮肤发红,粗糙、变硬,逐渐加重至增厚、皲裂、多汗而来诊。据患者自诉,出生不久便出现掌跖皮肤发红、薄而脆、粗糙、皲裂、变硬、手足多汗。皮肤科情况:掌跖明显弥漫性角化过度,境界清楚,色淡黄,表面光滑半透明、干燥、发硬,冬季可有皲裂现象(见图1、2),其女情况类似(见图3、4)。患者既往体健,发育正常,身体匀称,毛发、牙齿均正常,无智力障碍。父母非近亲婚配,家族中有类似疾病患者。自涂多种药膏(名不详) 无效,未到医院诊治过。
病理:表皮角化过度,颗粒层、棘层肥厚,形成嵴状,基底细胞层完整。真皮浅层血管周围少量淋巴、组织细胞浸润(图6、7)。
家系调查:该家系4代共30人,男13人,女17人,无近亲婚配,患病11例,其中男性5例,女性6例,见图5。整个家系中,患者均于幼年发病,患者年龄最小者2岁,最大者71岁(先证者外祖父)。
讨 论 掌跖角化症(palmoplantar keratoderma,PPK) 最初由德国医生Vorner于1901年报道,包括十几种类型,在临床上常见的类型是弥漫性掌跖角化症(dif-fuse epidemolytic palmopantar keratodemadiffuse EPPK)。按照stevens等(1996)所提出弥漫性掌跖角化症又可分为获得性弥漫型、表皮松解型、非表皮松解型和可变性红斑角皮病4种亚型。除获得性弥漫型外,其余3种亚型一般生后发病,持续终生,是一种常染色体显性遗传性疾病(OMIM:144200)[1]。本家系连续4代发病,男女均受累,符合常染色体显性遗传。表皮松解型是目前研究最多的一种弥漫性掌跖角化症,其致病基因(KRT9)定位于17q21.1-q21.2,蛋白质产物为角蛋白9(keratin 9),已发现十几种基因突变,目前尚无有效根治疗法和产前诊断手段。本病尚无有效的根治疗法和准确的产前诊断手段,对工作和生活有一定的影响[2]。患者一般为显性杂合子基因型,每生育一个孩子,都有1/2的发病风险。因此,医师应该劝阻患者明智选择放弃生育后代。
[1]赵辨.中国临床皮肤病学[M].江苏科学技术出版社,2010:1074-1077.
[2]王文强.掌跖角化皮肤综合征一家系的遗传学研究[J].中国优生与遗传杂志,2000,8(5):110.
猜你喜欢
父母必读(2023年9期)2023-09-24 12:59:03
小天使·五年级语数英综合(2021年12期)2021-03-21 00:46:59
家庭医药(2020年6期)2020-06-30 10:10:46
中国医药科学(2019年2期)2019-06-10 11:42:00
东方教育(2017年14期)2017-09-25 02:07:38
中国中医药现代远程教育(2017年9期)2017-01-28 16:05:59
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:54
贵州医科大学学报(2016年6期)2016-06-30 03:19:50
课程教育研究·学法教法研究(2016年1期)2016-03-17 15:52:58
西南医科大学学报(2015年1期)2015-08-22 13:02:00
