
氨硼烷水解制氢的研究进展
2014-02-13 09:26杨兰罗威程功臻
大学化学 2014年6期
杨兰 罗威程功臻
(武汉大学化学与分子科学学院 湖北武汉430072)
由于石油等化石燃料的长期大量消耗,石油资源逐渐枯竭,而人类对能源的需求量越来越高,新能源的开发成为各国发展的首要问题。氢能作为一种最为洁净的能源[1],被人们越来越重视。氢气的含能量为120MJ·kg-1,是石油的3倍;并且氢气燃烧后的产物是水,是最为清洁的能源。然而,制约氢能应用的最重要的因素是氢能的储存和运输问题。储氢材料(包括化学氢化物[2],吸附储氢材料[3],金属氢化物[4])近年来被广泛研究。在储氢材料中,硼氮化合物由于高的质量储氢密度和容易脱氢的特点而受到特别的关注。其中,氨硼烷具有19.6%的质量储氢密度,远远高于美国能源部的2017年5.5%的质量储氢目标[5]。氨硼烷具有高的稳定性和环境友好性,被作为一种有潜力的储氢材料。氨硼烷的脱氢方式有3种,分别为溶剂解[6]、热分解[7]和水解[1],其中,氨硼烷的水解是最方便的一种脱氢方式,能够在室温下,短时间内水解释放出3分子的氢气,反应式如式(1)所示。

氨硼烷的水溶液在室温下是非常稳定的,而在有金属催化剂的作用下,氨硼烷能够快速地水解放出氢气。氨硼烷水解的催化剂主要是过渡金属纳米粒子,包括贵金属和非贵金属催化剂,贵金属主要有Pd[8]、Rh[9]、Ru[10]和Pt[11],非贵金属主要为Fe[12]、Co[13]、Ni[14]和Cu[15]。本文从氨硼烷的催化剂入手,简要介绍近年来氨硼烷水解的研究进展。
1 氨硼烷的合成及表征
1.1 氨硼烷的合成
1955年,Shore和Parry首次合成出氨硼烷[16]。其反应式如式(2)所示。

这种方法是利用铵盐和金属硼氢化物反应合成氨硼烷,报道的收率为45%。为了提高收率,使用了不同的铵盐和金属硼氢化物对这种方法加以改进[17],反应通过硫酸铵和硼氢化钠在无水无氧条件下进行,反应溶剂为THF,反应温度为40℃。反应式如式(3)所示:

铵盐和硼氢化钠反应得到氨硼烷的反应还有很多,方法简单,如表1所示[18]。

表1 四氢呋喃中硼氢化钠和铵盐合成氨硼烷
1.2 氨硼烷的表征
氨硼烷通过X射线衍射(XRD),FT-IR,拉曼图和核磁共振光谱进行了表征。图1为氨硼烷的XRD图谱,氨硼烷具有四方晶体结构,特征峰在24°,34°,41°,晶胞参数如表2所示[19]。

表2 氨硼烷的晶胞参数

图1氨硼烷的XRD图谱
图2(a)为氨硼烷的傅立叶红外图,其中3308cm-1为N—H的伸缩振动峰,2276cm-1为B—H的伸缩振动峰,1598cm-1为N—H的弯曲振动峰,1372~1055cm-1为B—H的弯曲振动峰,1373cm-1为N—H的摇摆振动峰,782cm-1和725cm-1为B—N的伸缩振动峰。图2(b)为氨硼烷的拉曼图,其中3264cm-1为N—H的伸缩振动峰,2400~2354cm-1为B—H的伸缩振动峰,1600和1368cm-1分别为N—H和B—H的弯曲振动峰,1373cm-1为N—H的摇摆振动峰,784cm-1为B—N的伸缩振动峰。
13B NMR硼谱是氨硼烷结构的重要表征手段,图3为氨硼烷的13B NMR谱图[20],-22.3处的四重峰为BH3的特征峰。

图2 氨硼烷的FT-IR(a)和拉曼图(b)
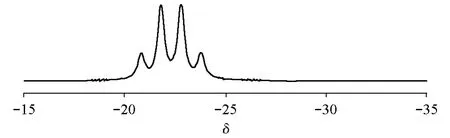
图3 氨硼烷的13B NMR图
2 催化剂
氨硼烷的水溶液在没有催化剂存在下是很稳定的。在室温下,空气中可以最多保存67天[21]。最早关于氨基硼烷水解的研究是在20世纪60年代[22-23],大多数研究的目的是确定氢的释放反应动力学。氨硼烷水解的催化剂主要为过渡金属催化剂,包括贵金属和非贵金属催化剂,为了降低成本,提高催化活性,有很多非贵金属掺杂的贵金属催化剂也被广泛的研究。
2.1 以贵金属为基础的催化剂
最早以过渡金属作为催化剂催化氨硼烷水解产氢的研究是2006年Chandra和Xu的工作[24],研究表明加入合适的金属催化剂如铂黑,钯黑,或铑前体催化剂([Rh(1,5-cod)(μ-Cl)]2),通过控制不同的催化剂量,可以使氨硼烷放出的氢气和NH3BH3的物质的量比达到3,相当于氢气的产量为起始原料NH3BH3和H2O的8.9%,超过了美国能源部5.5%的质量储氢目标。由于金属纳米粒子具有高的表面能容易发生聚集,从而造成催化活性的降低,为了提高催化剂的催化活性和稳定性,Xu等人[25]报道了由γ-Al2O3和SiO2负载的Ru、Rh、Pd、Pt、Au纳米颗粒催化氨硼烷水解的体系。后来又有更多的材料被用来作为金属纳米颗粒的载体,常用的有炭黑[26],聚乙烯吡咯烷酮(PVP)[27],石墨烯[28]等。Ma等人[26]报道了在炭黑上负载的金属钌催化剂Ru/C,反应时间仅需65s,具有目前催化氨硼烷水解最高的TOF值(429.5mol(H2)·min-1·mol(Ru)-1)。尽管贵金属具有很高的催化活性,但是要实际应用的成本相对还是很高。为了降低催化剂成本,在贵金属中掺杂非贵金属是一种很好的选择,所掺杂的非贵金属主要为过渡金属中的Fe[29],Co[30],Ni[31]和Cu[32]等。我们组报道了负载在炭黑上的金属钌基催化剂Ru@Co/C和Ru@Ni/C[29],都具有很好的催化氨硼烷水解的反应活性,完全反应时间分别为160s和200s,对于贵金属Ru而言,TOF值分别为319.7和250.1mol(H2)·min-1·mol(Ru)-1,这样不仅具有很高的反应活性,也大大降低了成本。
石墨烯是单原子厚度的碳原子层,具有良好的导热性[33]、电学性能[34],以及大的比表面积[35],是一种理想金属纳米颗粒载体。近年来,用石墨烯负载的金属纳米催化剂广泛应用于氨硼烷的水解中[36-37]。RGO/Pd催化剂[37]是通过一步法将氧化石墨烯和金属离子还原得到的,该催化剂具有很好的催化活性和循环活性,5次循环后活性还有初始的65%。另外一种合成石墨烯负载的催化剂的方法是先将氧化石墨烯通过还原剂还原,然后将金属离子还原为金属纳米颗粒,最后将纳米颗粒负载在还原得到的石墨烯上。CDG/Pd催化剂[37]就是通过这种方法得到的。石墨烯是通过水合肼还原得到的;金属纳米粒子是通过叔丁基胺硼烷(BTB)作为还原剂,在油胺体系中还原得到的,这种方法得到的是单分散的纳米颗粒;然后将这些纳米颗粒和石墨烯在正己烷中搅拌过夜,得到CDG/Pd催化剂(图4)。
我们课题组做了很多关于石墨烯负载的金属纳米颗粒催化剂用于氨硼烷水解方面的研究,通过和上面类似的以油胺作为稳定剂的方法,得到了Ag@Co/RGO催化剂[28]。更多的是,我们用还原性较弱的氨硼烷或甲基氨硼烷作为还原剂,选择性地先将电势较高的贵金属还原形成核种子,再诱导非贵金属在核种子表面还原,通过一步法得到石墨烯负载的核壳结构纳米催化剂;以往合成核壳结构的纳米颗粒的方法为先还原一种金属离子,再加入另外一种或多种金属和还原剂,将还原得到壳。我们所使用的方法是一步法合成核壳结构,方法简单易操作,在室温下有很好的催化氨硼烷水解的催化活性。以金属钌为基础的催化剂有Ru/RGO[38],Ru@Co/RGO[39]和Ru@Ni/RGO[40],其中Ru/RGO颗粒的大小为1.1±0.1nm,水解氨硼烷反应的活化能为11.7kJ/mol,是目前报道的氨硼烷水解反应中活化能最小的。除了金属钌以外,我们还研究了金属银为基础的核壳结构的合成,主要有双核的Ag@Co/RGO、Ag@Ni/RGO[28],以及三核的Ag@CoNi/RGO[41]、Ag@CoFe/RGO和Ag@NiFe/RGO[42]。用这种简单的方法也可以得到分散性很好的纳米颗粒,如图5所示为Ru/RGO和Ag@Co/RGO的TEM图,纳米颗粒均匀地分散在石墨烯上,可以有效防止纳米粒子的聚集。这种催化剂不仅具有很好的催化氨硼烷水解的活性,也能催化甲基氨硼烷的水解,反应活性也很高,能够在10min左右催化甲基氨硼烷的水解反应完全进行。在此之前,甲基氨硼烷的水解还没有被研究过,甲基氨硼烷具有11.1%的理论储氢量,是一种很有潜力的储氢材料。一些已报道的以贵金属为基础氨硼烷水解脱氢的催化剂的TOF值和活化能总结在表3中。

图4 油胺稳定的Pd纳米颗粒的TEM图(a)和CDG/Pd的TEM图(b)
2.2 非贵金属催化剂
尽管贵金属在催化氨硼烷水解中表现出了非常高的催化活性,但是从实际应用的角度来看,催化剂应该具备高效率、低成本、稳定性好和反应条件温和等特性。所以使用非贵金属催化剂催化氨硼烷水解脱氢是十分必要的。Xu等[13]首先研究了非贵金属催化剂在室温下对氨硼烷水解的催化性能,发现Co、Ni和Cu都能使氨硼烷完全水解放出3分子的氢气,而Fe则不能。从图6可以看出,Co/γ-Al2O3具有最好的催化效果(图6(a));此外,不同载体的Co的催化剂Co/C和Co/SiO2,都能在1h内催化氨硼烷完全水解(图6(b)),这种催化剂的合成是先将金属硝酸盐的水溶液和载体充分搅拌,将水蒸发出去,然后将得到的样品在空气中煅烧,再在氢气流下高温还原得到。这种方法费时费力,为了进一步简化催化剂的合成方法和加快催化反应的速度,又有很多课题组对非贵金属的催化剂进行了大量的研究。

图5 Ru/RGO(a)和Ag@Co/RGO催化剂的TEM图(b)

表3 以贵金属为基础的氨硼烷水解脱氢的催化剂的催化活性

图6 (a)由γ-Al2O3负载的催化剂催化NH3BH3水溶液(1%,10mL)水解的脱氢图;(b)不用载体的Co催化剂的脱氢图,其中,n(金属)/n(NH3BH3)=0.018。
Li等人[57]通过水热法得到了具有优异催化性能的Co-MOF催化剂。从图7中可以看出,Co-MOF催化剂能够在2min内完全催化氨硼烷的水解,并且具有很好的稳定性,在空气中放置30min仍然能够在4min内反应完全。
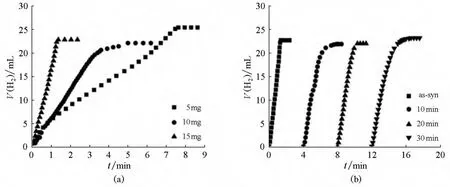
图7 (a)不同量的催化剂催化氨硼烷的水解(0.32mol/L,1.0mL);(b)催化剂在空气中放置不同时间后的催化性能,反应在室温下进行。
Patel等人[58]合成了3种介孔二氧化硅负载的Co-B催化剂,Co-B/MCM-41、Co-B/FSM-16和Co-B/SBA-15,在室温下进行对氨硼烷水解反应的催化,加入负载后的催化活性比没有负载的活性明显有所提高,其中Co-B/SBA-15具有最好的催化活性。Özkar等人[59]研究了羟基磷灰石(HAP)负载的Co纳米簇催化剂Co(0)-HAP,这种催化剂具有好的循环性能,5次循环后还有81%的催化活性;催化活性的降低可能是因为水解过程中得到了偏硼酸盐,增大了反应体系的黏度,从而降低了反应活性。主要的单金属催化剂是金属Ni催化剂,包括中空的Ni-SiO2[14],PSMA-Ni[60],Ni-ZIF-8[61],Ni-PVP[27]等。为了进一步提高催化剂的反应活性,有很多关于双金属,三金属的催化剂的研究。Lu等人[62]合成了聚乙烯亚胺修饰的氧化石墨烯负载的铁镍合金催化剂PEI-GO/Fe-Ni,首先是合成了PEI-GO,然后将PEI-GO和硫酸亚铁和硝酸镍的水溶液混合,加入强还原剂NaBH4,通过强烈震荡还原得到,这是典型的合成金属合金的方法。这种催化剂的优点在于具有磁性,能够通过磁铁很容易地从反应体系中分离出来,使工业应用在很大程度上成为了可能。图8说明了这种催化剂的磁性。类似有磁性的催化剂还有很多,如CuNPs@SCF[15]、PEG稳定的Fe(0)纳米簇[12]、Fe0.5Ni0.5纳米催化剂[63],CoNi/RGO[64]等。

图8 放置磁铁之前(a)和之后(b)
用硼氢化钠还原可以得到金属合金,用较弱的还原剂如氨硼烷和甲基氨硼烷可以还原得到核壳结构,我们课题组用甲基氨硼烷作为还原剂合成了Cu@Co/RGO[65]和Cu@CoNi/RGO[66]催化剂,合成机理见图9,具有较高电极电势的Cu2+首先被还原,Cu0诱导Co2+的还原,同时GO被还原得到RGO。其他关于银基和钌基核壳也是用同样的机理得到的。

图9 Cu@Co/RGO合成过程中颜色的变化(a)及机理图(b)(灰色平板代表GO,黑色平板代表RGO)。
一些已报道的非贵金属催化氨硼烷水解脱氢的催化剂的TOF值和活化能总结在表4中。

表4 非贵金属的氨硼烷水解脱氢的催化剂的催化活性
3 总结与展望
氨硼烷由于具有高的理论含氢量而被作为一种很有潜力的储氢材料;氨硼烷的水解能够在催化剂的催化下(室温)快速释放出3分子氢气。近年来关于氨硼烷的水解的研究主要集中在合成稳定、催化活性高、循环性能好、成本低、易回收、合成方法简单的催化剂,为氨硼烷的实际应用提供了可能性。但是,要在这些性质里面找到一个平衡点,得到最优的催化剂是目前的研究热点和难点。另外,氨硼烷水解制氢后的产物(偏硼酸盐)的合理回收再生,也是目前急需解决的问题。
[1]Jiang H L,Xu Q.Catal Today,2011,170:56
[2]James Jr C W,Brinkman K S,Gray J R,et al.Ⅰnt J Hydrogen Energy,2014,39:1371
[3]Attia N F,Lee S M,Kim H J,et al.Ⅰnt J Energy Res,2014,38:466
[4]Oumellal Y,Courty M,Rougier A,et al.Ⅰnt J Hydrogen Energy,2014,39:1
[5]Tang C M,Chen S W,Zhu W H,et al.Chem Phy Lett,2013,586:116
[6]Fortman G C,Slawin A M Z,Nolan S P.Organometallics,2011,30:5487
[7]Huang Z G,Lingam H K,Chen X N,et al.RSC Adv,2013,3:7460
[8]Wang J,Qin Y L,Liu X,et al.J Mater Chem,2012,22:12468
[9]Durap F,Zahmakɩran M,Özkar S.Appl Catal A:General,2009,369:53
[10]Liang H Y,Chen G Z,Desinan S,et al.Ⅰnt J Hydrogen Energy,2012,37:17921
[11]Chandra M,Xu Q.J Power Sources,2007,168:135
[12]DinɕM,MetinaÖ,Özkar S.Catal Today,2012,183:10
[13]Xu Q,Chandra M.J Power Sources,2006,163:364
[14]Umegaki T,Yan J M,Zhang X B,et al.J Power Sources,2009,191:209
[15]Kaya M,Zahmakiran M,Özkar S,et al.ACS Appl MaterⅠnterfaces,2012,4:3866
[16]Shore S G,Parry R W.J Am Chem Soc,1955,77:6084
[17]Yamada Y,Yano K,Fukuzumi S.Energy Environ Sci,2012,5:5356
[18]Ramachandran P V,Gagare P D.Ⅰnorg Chem,2007,46:7810
[19]Figen A K,Pişkin M B,Coşkuner B.Ⅰnt J Hydrogen Energy,2013,38:16215
[20]Daly S R,Bellott B J,Kim D Y,et al.J Am Chem Soc,2010,132:7254
[21]Brockman A,Zheng Y,Gore J.Ⅰnt J Hydrogen Energy,2010,35:7350
[22]Ryschkewitsch G E.J Am Chem Soc,1960,82:3290
[23]Kelly H C,Marchelli F R,Giusto M B.Ⅰnorg Chem,1964,3(3):431
[24]Xu Q,Chandra M.J Power Sources,2006,163:364
[25]Chandra M,Xu Q.J Power Sources,2007,168:135
[26]Liang H Y,Chen G Z,Desinan S,et al.Ⅰnt J Hydrogen Energy,2012,37:17921
[27]Yang L,Luo W,Cheng G Z.Catal Lett,2013,143:873
[28]Yang L,Luo W,Cheng G Z.ACS Appl MaterⅠnterfaces,2013,5:8231
[29]Cao N,Su J,Hong X L,et al.Chem Asian J,2014,9:562
[30]Barakat N A M.Appl Catal A:General,2013,451:21
[31]Aranishi K,Singh A K,Xu Q.ChemCatChem,2013,5:2248
[32]Cao N,Hu K,Luo W,et al.J Alloys Comp,2014,590:241
[33]Xu X D,Gabor N M,Alden J S,et al.Nano Lett,2010,10:562
[34]Wang L,Chen Z Y,Dean C R,et al.ACS Nano,2012,6:9314
[35]Shmavonyan G H,Sevoyan G G,Aroutiounian V M.Armen J Phy,2013,6:1
[36]Xi P X,Chen F J,Xie G Q,et al.Nanoscale,2012,4:5597
[37]MetinÖ,Kayhan E,Özkar S,et al.Ⅰnt J Hydrogen Energy,2012,37:8161
[38]Cao N,Luo W,Cheng G Z.Ⅰnt J Hydrogen Energy,2012,38:11964
[39]Cao N,Su J,Luo W,et al.Catal Commun,2014,43:47
[40]Cao N,Su J,Luo W,et al.Ⅰnt J Hydrogen Energy,2014,39:426
[41]Yang L,Su J,Meng X Y,et al.J Mater Chem A,2013,1:10016
[42]Yang L,Su J,Luo W,et al.Ⅰnt J Hydrogen Energy,2014,39:3360
[43]Akbayrak S,Özkar S.ACS Appl MaterⅠnterfaces,2012,4:6302
[44]Yang X J,Cheng F Y,Liang J,et al.Ⅰnt J Hydrogen Energy,2011,36:1984
[45]Yang X J,Cheng F Y,Tao Z L,et al.J Power Sources,2011,196:2785
[46]Aijaz A,Karkamkar A,Choi Y J,et al.J Am Chem Soc,2012,134:13926
[47]Chen G Z,Desinan S,Rosei R.Chem Eur J,2012,18:7925
[48]MetinÖ,ŞxahinŞ,Özkar S.Ⅰnt J Hydrogen Energy,2009,34:6304
[49]Chen G Z,Desinan S,Nechache R,et al.Chem Commun,2011,47:6308
[50]Can H,MetinÖ.Appl Catal B:Environ,2012,125:304
[51]Durap F,Zahmakɩran M,Özkar S.Ⅰnt J Hydrogen Energy,2009,34:7223
[52]Rachiero G P,Demirci U B,Miele P.Ⅰnt J Hydrogen Energy,2011,36:7051
[53]Rachiero G P,Demirci U B,Miele P.Catal Today,2011,170:85
[54]Sun B L,Wen M,Wu Q S,et al.Adv Funct Mater,2012,22:2860
[55]Xi P X,Chen F J,Xie G Q,et al.Nanoscale,2012,4:5597
[56]Wen M,Sun B L,Zhou B,et al.J Mater Chem,2012,22:11988
[57]Song P,Li Y Q,Li W,et al.Ⅰnt J Hydrogen Energy,2011,36:10468
[58]Gupta S,Patel N,Fernandes R,et al.AⅠP Conf Proc,2013,1512:284
[59]Rakap M,Özkar S.Catal Today,2012,183:17
[60]MetinÖ,Özkar S.Ⅰnt J Hydrogen Energy,2011,36:1424
[61]Li P Z,Aranishi K,Xu Q.Chem Commun,2012,48:3173
[62]Zhou X H,Chen Z X,Yan D H,et al.J Mater Chem,2012,22:13506
[63]Yan J M,Zhang X B,Han S,et al.J Power Sources,2009,194:478
[64]Feng W Q,Yang L,Cao N,et al.Ⅰnt J Hydrogen Energy,2014,39:3371
[65]Du Y S,Cao N,Yang L,et al.New J Chem,2013,37:3035
[66]Meng X Y,Yang L,Cao N,et al.ChemPlusChem,2014,79:325
猜你喜欢
分子催化(2022年1期)2022-11-02
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
无机盐工业(2021年9期)2021-09-09
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
信息记录材料(2016年4期)2016-03-11
互联网天地(2015年1期)2015-12-04
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年9期)2014-02-28
无机化学学报(2014年4期)2014-02-28
