
高效液相色谱法测定蒙成药升阳十一味丸中没食子酸含量*
2014-01-19 07:57郭宝凤王玉华侯海玲
中国药业 2014年20期
郭宝凤,王玉华,侯海玲
(内蒙古医科大学药学院,内蒙古 呼和浩特 010110)
升阳十一味丸由全石榴、益智、红花、荜茇、黄精、玉竹、蒺藜、肉桂、天花粉、冬葵果、天冬11 味药材组方,具有暖肾、利水、消食、燥“协日乌素”的功效,可用于胃寒、消化不良、肾寒腰疼、寒性腹泻等症,临床应用广泛。其药品标准收载于1998 年版《中华人民共和国卫生部药品标准·蒙药分册》[1],现有质量标准中没有含量测定内容。笔者建立了测定升阳十一味方中君药全石榴没食子酸含量的高效液相色谱(HPLC)法,现报道如下。
1 仪器与试药
日本L-2000 型高效液相色谱仪,DAD 检测器,EZChrom Elite for Hitachi Version 工作站;Mettle RAE240 型电子天平(梅特勒-托利多有限公司);28HVLRASOTH 型超声处理器(昆山市超声仪器有限公司)。升阳十一味丸(编号A,B,C,内蒙古乌兰浩特中蒙制药有限公司,批号分别为090214,090701,090313;编号D,E,F,阜新蒙药有限责任公司,批号分别为20100302,20060224,20100414;编号G,内蒙古蒙药股份有限公司,批号为100537);没食子酸对照品(中国食品药品检定研究院,批号为110831 -200302,供含量测定用);甲醇为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 溶液制备
称取没食子酸对照品适量,精密称定,置容量瓶中,加甲醇制成每1 mL 含没食子酸27 μg 的溶液,即得对照品溶液。取样品粉末约0.5 g,精密称定,置具塞锥形瓶中,精密加入75%乙醇25 mL,超声处理(功率220 W,频率40 kHz)30 min,过滤,滤液50 ℃减压旋干,用25 mL 水分次转入60 mL 分液漏斗中,用醋酸乙酯萃取5 次,每次25 mL,合并萃取液,30 ℃减压旋干,残渣用甲醇分次溶解,转移至25mL 容量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得供试品溶液。按照处方比例及制备工艺,取缺全石榴的方中其他药材粉末,制备阴性对照品,精密称取适量,按供试品溶液制备方法制成阴性对照品溶液。
2.2 色谱条件
色谱柱:菲罗门Kromasil C18柱(250 mm×4.6 mm,5 μm);流动相:甲醇-0.1%磷酸水溶液(5 ∶95);检测波长:273 nm;流速:1.0 mL/min;柱温:30 ℃;进样量:10 μL。理论板数按没食子酸的峰计算不低于4 000。
2.3 方法学考察
专属性试验:分别吸取对照品溶液、供试品溶液和阴性对照品溶液各10 μL,注入液相色谱仪,按拟订色谱条件进样测定,色谱图见图1。在没食子酸对照品色谱峰的保留时间处,供试品溶液有色谱峰且与相邻峰得到基线分离,阴性照品溶液在此处无色谱峰,说明共存的其他药味对测定无干扰。
线性关系考察:精密称取没食子酸对照品1.36 mg,置50 mL容量瓶中,加甲醇溶解并稀释至刻度(质量浓度为27.02 μg/mL),精密吸取2,5,10,15,20 μL,分别注入液相色谱仪中,按拟订色谱条件测定,记录色谱图。以进样量(X,μg)对峰面积积分值(Y)进行回归分析,得回归方程Y=12 095 X-797 509,r=1.000 0(n=5)。结果表明,没食子酸进样量在54.04 ~540.4 ng 范围内与峰面积积分值呈良好线性关系。
精密度试验:取同一没食子酸对照品溶液(质量浓度为27.02 μg/mL),进样10 μL,连续测定5 次。结果峰面积平均值为2 188 165,RSD=1.35%(n=5),表明仪器精密度良好。
稳定性试验:取同一供试品(批号为090701)溶液,分别于制备后0,2,4,6,8,12,24 h 时进样测定。结果的RSD=1.89%(n=7),表明供试品溶液在24 h 内基本稳定。
重复性试验:取同一批(批号为090701)供试品6 份,每份取样量约0.5 g,精密称定,依法制备供试品溶液进样测定。结果平均含量为1.288 mg /g,RSD =1.90%(n =6),表明方法重复性良好。
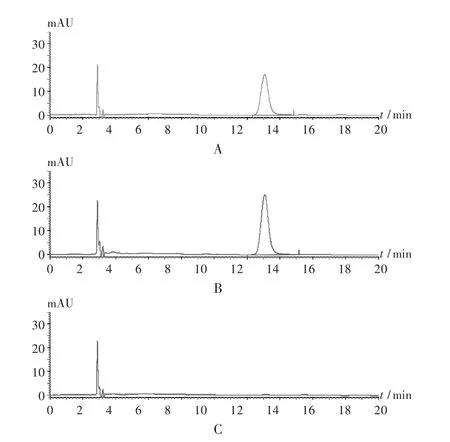
图1 专属性试验高效液相色谱图
加样回收试验:取已知含量的供试品(批号为090701,没食子酸含量为1.288 mg/g)粉末约0.25 g,精密称定,共9 份,分别加入没食子酸对照品0.259 4,0.324 2,0.389 1 mg,各3 份。分别按供试品溶液制备方法制备溶液,按拟订色谱条件测定,记录色谱图,计算回收率。结果见表1。
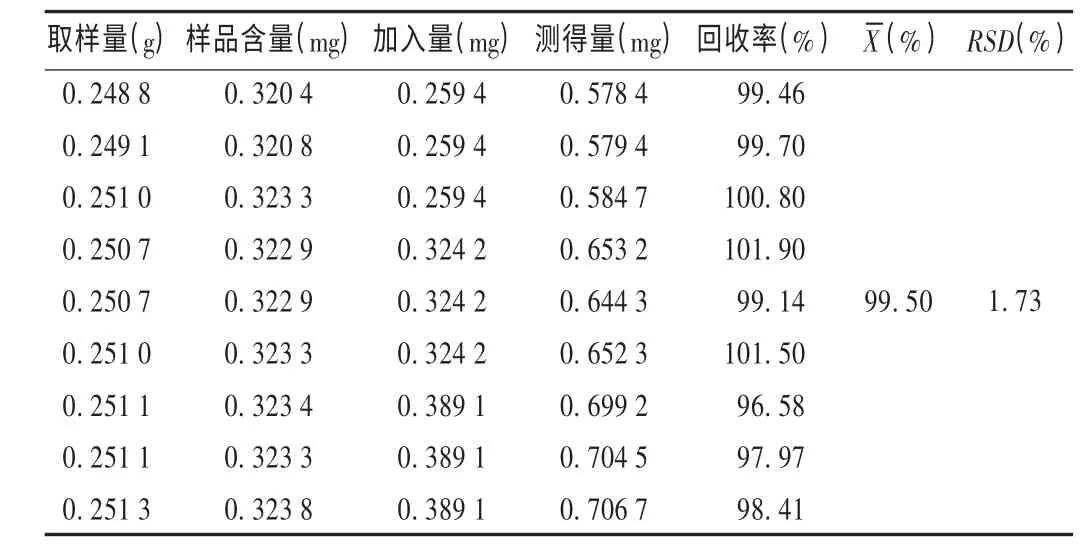
表1 没食子酸加样回收试验结果(n=9)
2.4 样品含量测定
取7 个批次的供试品各0.5 g,依法制备供试品溶液,进样10 μL,按拟订色谱条件测定,计算含量。结果见表2。可见,样品中没食子酸的含量为0.5344 ~1.349 mg/g,不同厂家之间存在差异,相同厂家的不同批次间也有差异。因此,对君药全石榴中没食子酸进行含量测定具有现实意义。
3 讨论
流动相的选择:2010 年版《中国药典(一部)》[1]及文献[2-3]测定没食子酸所采用的流动相为不同比例的甲醇-磷酸水溶液和乙腈-0.1%磷酸水溶液(含0.1%三乙胺)。经试验筛选,选择甲醇-0.1%磷酸水溶液(5 ∶95)为流动相。
检测波长的选择:2010 年版《中国药典(一部)》及文献[4-7]中采用HPLC 法测定单药材及成方制剂中没食子酸的含量,所用检测波长有216,273,267,275 nm。本试验中,在200 ~800 nm 波长范围内对没食子酸对照品溶液进行光谱扫描。结果光谱图中有2 个吸收峰,波长分别为220,273 nm。考虑在220 nm 波长为末端吸收、干扰较多,273 nm 波长处的峰高且相对较锐,故选择273 nm为测定波长。
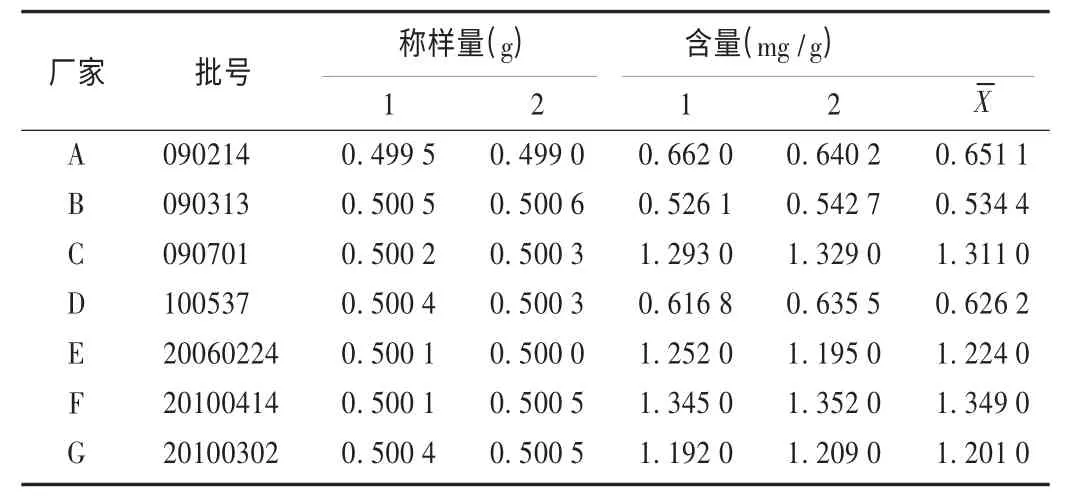
表2 样品含量测定结果(n=2)
超声时间的选择:分别考察了不同超声提取时间对没食子酸含量的影响。结果表明,超声30 min 与20 min 比较,随着超声时间的延长,含量明显提高;超声30 min 与40 min 比较,两者含量没有明显的变化。故确定超声时间为30 min。
萃取次数的选择:该方由全石榴等10 味药组方,全石榴为君药,没食子酸又为其主要成分[8],故以没食子酸为指标性成分进行含量测定[9]。但含量测定时干扰成分较多,故在前处理时增加了萃取步骤。选用乙酸乙酯进行萃取并对萃取次数进行考察,结果表明,随着萃取次数的增加,没食子酸含量增加;萃取5 次与萃取4 次的结果比较,前者的含量明显增高,但与萃取6 次的结果比较时含量没有明显变化。故确定用乙酸乙酯萃取5 次。
柱耐用性试验:为了考察不同ODS 柱(型号和柱效均不同)对测定结果的影响,选用3 种型号的色谱柱对同一供试品(批号为20100414)溶液进行测定。结果显示,没食子酸峰与相邻峰均能达到基线分离,峰对称性均好,且各柱含量测定结果之间没有明显差异,说明色谱柱理论塔板数在3 000 以上时柱型号对结果没有影响。
[1] 国家药典委员会. 中华人民共和国药典(一部)[M]. 北京:中国医药科技出版社,2010:173.
[2] 霍 文,刘占军,张婷婷. 反相高效液相色谱法测定石榴皮中没食子酸的含量[J]. 西北药学杂志,2007,22(5):242 -243.
[3] 王克英. 石榴皮中没食子酸定性定量方法研究[J]. 中国中药杂志,2005,30(15):1 171 -1 172.
[4] 许 勇,郏征伟,诸艳蓉,等.HPLC 法测定扎冲十三味丸中没食子酸的含量[J]. 中国卫生检验杂志,2012,22(7):1 560 -1 561.
[5] 霍生青.HPLC 测定十三味菥冥丸中的没食子酸[J]. 华西药学杂志,2012,27(4):100 -101.
[6] 张朔生.HPLC 测定炮制前后石榴皮中没食子酸的含量[J]. 药物分析杂志,2010,30(6):137 -139.
[7] 林 夏,胡军华,崔培超,等.HPLC 同时测定大花红景天提取物中没食子酸、红景天苷、酪醇、对香豆酸的含量[J]. 中国实验方剂学杂志,2013,10(19):102 -105.
[8] 张 倩,杜海云,陈令梅,等. 石榴化学成分及其生物活性研究进展[J]. 落叶果树,2010(6):17 -22..
[9] 刘 源,沈好文. 高效液相色谱法测定化痔灵片中没食子酸含量[J].中国药业,2013,22(8):56 -58.
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
音乐天地(音乐创作版)(2021年7期)2021-10-13
阅读(科学探秘)(2021年8期)2021-09-01
草原歌声(2021年1期)2021-07-16
少先队活动(2021年1期)2021-03-29
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
小天使·一年级语数英综合(2018年4期)2018-06-22
中国医疗美容(2015年4期)2015-04-27
中华皮肤科杂志(2014年3期)2014-12-19
