
单链环状DNA添加剂抑制基因表达序列分析Ditag聚合酶链反应引物多聚体生成
2014-01-18 14:03刘玉慧刘星刘学东郑冬
安徽农业科学 2014年15期
刘玉慧,刘星,刘学东,郑冬
(东北林业大学,黑龙江哈尔滨 150040)
单链环状DNA添加剂抑制基因表达序列分析Ditag聚合酶链反应引物多聚体生成
刘玉慧,刘星,刘学东,郑冬*
(东北林业大学,黑龙江哈尔滨 150040)
[目的]研究利用单链环状DNA抑制聚合酶链反应(PCR)副产物的作用效果。[方法]设计并合成单链环状DNA,并以具同样序列发卡状结构的单链DNA为对照组,在基因表达序列分析(SAGE)Ditag PCR反应中加入上述不同浓度的DNA,利用电泳技术检测不同结构(环状/单链)以及不同浓度单链DNA对PCR多聚体附产物的抑制作用。[结果]发卡状结构的单链DNA无法抑制PCR引物多聚体副产物的产生;而在70~150 nmol/L终浓度范围内,单链环状DNA可以抑制SAGE Ditag PCR反应中引物多聚体的生成,并且不影响SAGE不同tag间的表达丰度差异。[结论]单链环状DNA可以作为PCR反应中引物多聚体生成的有效抑制剂。
单链环状DNA(sscDNA);系列基因表达分析(SAGE);引物多聚体;抑制
随着基因测序技术的不断完善和发展,许多模式物种和高等动物(如人)的基因组全序列的信息被一一破译。然而,基因组计划的研究成果只能从结构水平说明基因的组成特征和序列信息;从功能水平来如何解读这些基因调控和相互作用成为后基因组时代一个亟待解决的问题[1]。例如,人大约有30 000个基因,但是大约只有20%的基因在同一时间被表达。基因表达的时空性已经引起广泛的关注。针对此问题,各种组学(Omics)水平的技术方法和理论应运而生的。其中从转录水平阐明在特定时间和空间节点中基因组表达网络的转录组学是近年来一个热点研究领域[2]。
目前常用的研究基因差异表达的试验方法有很多种。常见的有mRNA差显RTPCR法(mRNA differential display RT-PCR,mRNA DDRT-PCR)、消减杂交(subtractive hybridization)等[3]。然而这些方法信息容纳量不足,仅能反映有限基因的表达水平,不能很好地揭示那些大量未知基因的表达水平。1995年美国Johns Hopkins大学的Velculescu等首先提出了系列基因表达分析(Serial analysis of gene expression,SAGE)技术,用于基因转录表达谱的分析。SAGE技术[4]的出现是组学研究上的一个重要里程碑,该技术能同时定量分析实验对象的成千上万个转录本,克服了以前很多技术存在的缺陷,还能很灵敏地检测出那些低丰度表达的基因。SAGE技术已经从纯粹的基因差异表达研究拓展到重要基因功能研究、医学基础理论研究、疾病的诊断和治疗等很多领域,并取得了诸多显著的成果[5]。
众所周知,理论上一个9个碱基的短核苷酸序列标签能够分辨262 144个不同的转录物(49),而人类基因组估计仅能编码约80 000种转录物,因此一个SAGE tag包含有足够的信息能够确认唯一一种转录物。在实际运用中,SAGE技术始终在不断改进和发展。以其中Long SAGE为例,其采用特定的锚定酶能获得约17 bp的SAGE标签(tag)来特异性地确定mRNA转录物,然后利用酶将每2个tag连接形成ditag,进行PCR扩增后,再将酶切后的将多个ditag标签(20~60个)随机串联形成大量的多联体(concatemer)并克隆到载体中;经测序并应用SAGE软件分析,可确定表达的基因种类,并可根据标签出现的频率确定基因的表达丰度[6]。由于SAGE是一个高度敏感系统,可用于低丰度序列的确定。目前该技术已经广泛应用于很多疾病相关新基因的发现,以及细胞或组织的基因表达谱的研究。
近年来,试验尝试利用SAGE技术研究鹿茸再生过程中基因表达网络的时空变化动态。在试验过程中发现,对SAGE ditag进行PCR扩增过程中有大量引物多聚体出现。由于PCR扩增SAGE ditag是SAGE技术的关键环节,通过这一步既要保证各tag能反映原始表达丰度的差异,同时还要获得足够量的DNA用于下游酶切反应;而引物多聚体对实现上述实验目标产生了极大地干扰。Brownie等曾研究通过设计新型引物来抑制PCR引物多聚体试验方案[7]。由于这将涉及对Ditag两端接头序列进行较大改动,而且需要2次PCR扩增,这显然不符合SAGE技术中关于保持表达丰度的基本原则。但是这个技术方法有一个启示:既然具有发卡结构的引物能够增加PCR目标产物的特异性合成,能否设计具有发卡结构的单链DNA作为添加剂来抑制PCR引物多聚体的产生呢?笔者针对这个设想开展试验研究,以期为该技术的进一步开发利用提供依据。
1 材料与方法
1.1 材料 SV Total RNA Isolation System,购自Promega公司;Long SAGE文库构建试剂盒,购自Invitrogen公司;SYBR Premix ExTaq试剂盒、Agarose ms-6、Agarose LM SIEVE,Ex taq和hot-start taq DNA polymerase,均购自Takara公司;单链DNA寡聚核苷酸(C1:GCGGGCGGCG CAAAAAAGCG CCGCCCGC和C2:CCCGCAAAAA AGCGGGCGGC GCAAAAAAGC GCCG)添加物、STAT1引物(S1:TGGGGCACAA GGTGACAG和S2:TCACTCTTCTGTGTTCACTTACACTTCA)以及人GAPDH实时定量PCR引物(HG1:AGAAGGCTGG GGCTCATTTG和HG2:AGGGGCCATC CACAGTCTTC),均由上海英骏公司合成与纯化,其中C1通过复性形成发卡结构;C2通过连接形成单链环状结构。
1.2 鹿茸上皮组织总RNA的提取 取鹿茸新鲜上皮组织30mg,放入1.5 ml离心管中。根据SV Total RNA Isolation System试剂盒试验手册,加入RNA Lysis Buffer后,将组织匀浆,进行总RNA的提取。用紫外分光光度计测定RNA在260和280 nm的OD值,计算浓度与纯度。取其中10μl总RNA和15μl甲酰胺65℃、5 min处理后迅速放到冰上冷却2 min。再利用1.2%的琼脂糖凝胶电泳分析总RNA的完整性。将获得的总RNA分装,保存于-70℃备用。其他总RNA保存于-70℃备用。
1.3 SAGE文库Ditag制备 根据Long SAGE文库构建试剂盒相关试验手册。取100μl oligo(dT)磁珠置于1.5 ml离心管中,在磁座上对oligo(dT)磁珠进行清洗2次后,将准备好的100μg RNA与磁珠进行充分混合。随后室温静置30 min,在磁座上吸附2 min,移去上清液;再用1×First Strand Buffer清洗4次,去除上清液;然后加入87μl cDNA一链合成液(18.0 μl 5 × First Strand Buffer;1.0 μl RNaseOUT;54.5 μl DEPC 水;9.0 μl0.1mol/LDTT;4.5 μl dNTPMix),37 ℃温育1 h进行cDNA第1链的合成。接着加入660μl cDNA 2链合成液(465μl DEPC Water;150μl 5×Second Strand Buffer;15 μl dNTPMix;5 μlE.coliDNA Ligase;20 μlE.coliDNA Polymerase;5μlE.coliRNase H),16℃温育反应2 h后置于冰上,并加入45μl0.5mol/L EDTA终止反应。接着加入750μlWash Buffer C 75℃温育12 min,灭活E.coliDNA Polymerase。在磁座上静置2min,移去上清液,用Wash Buffer D洗脱4次。最后,吸去上清,加入200μl 1×Buffer 4,摇匀。
在磁座上静置2 min后,去除上清,将磁珠悬浮于200μl NlaⅢ酶切溶液中(172μl LoTE;2μl 100×BSA;20μl 10×Buffer 4;6μl NlaⅢ),37℃温育1 h后,将反应液在磁座上静置2min,吸去上清液,用750μlWash Buffer C洗脱并灭活酶的活性,再用Wash Buffer D洗脱磁珠4次。用150μl 1×Ligase Buffer洗脱2次,随后用100μl 1×Ligase Buffer悬浮磁珠,等量分成A、B管。
将上述2管溶液静置在磁座上2 min,小心移去上清液,逐步加入接头连接液(1.5μl Adapter A或B(20 ng/μl);14.0 μl LoTE;2.0 μl 10×Ligase Buffer);50 ℃加热2 min,室温冷却15 min,放置于冰上。随后加入2.5μl T4 DNA连接酶,16℃温浴2 h。每1管都分别用500μlWash Buffer D洗脱4次,再用1×Buffer4洗2次,将磁珠悬浮在1×Buffer4溶液中。
将上面得到的2管溶液静置在磁座上2min后移去上清液。加入含有标签酶的反应液(70μl LoTE;10μl 100×SAM;10μl10×Buffer4);37℃温浴2min,平衡反应液,加入10μl标签酶MmeI,37℃温浴1 h,反应结束后,在磁座上静置2 min,小心吸取上清液,分别保存于新的2个离心管中。利用酚-氯仿及乙醇沉淀法处理所得到的样品,分别用4μl LoTE溶解沉淀。将上述2管溶液混合,加入连接酶及反应液,37℃温浴30 min后。利用酚/氯仿及乙醇沉淀法处理所得到的Ditag,最后的沉淀颗粒溶解在14μl LoTE溶液中。
1.4 SAGE Ditag的扩增 将连接产物40倍作为PCR扩增模板,进行Ditag PCR反应。试验每个反应体系(50μl)组成均如下:5 μl 10 × PCR buffer,3 μl DMSO,5 μl dNTP,0.5 UTaqDNA聚合酶,SAGE上下游引物各2μl。其他反应成分如不同浓度单链DNA添加剂和DNA或cDNA模板因试验不同目的不同均为1μl或用等体积水补足。PCR反应条件为:2min,95 ℃进行变性;94 ℃,30 s;55 ℃,1 min;70 ℃,1 min;共计27个循环进行扩增;最后70℃延伸5min。所有试验均设阴性和/或阳性对照组。PCR扩增产物用4%琼脂糖凝胶电泳检测。
2 结果与分析
2.1 单链环状DNA添加剂可抑制引物多聚体 试验使用SAGE文库构建试剂盒中的引物进行ditag扩增时,引物可自身产生2个以上的清楚可见的多聚体DNA带,虽然使用试剂盒附带的热启动DNA聚合酶,但是无法抑制上述副产物的产生(图1A),这将影响ditag目的带的扩增效率。试验发现,其中一条引物多聚体带与真正的130 bp ditag带非常接近,对于ditag带切胶回收具有干扰。合成单链DNA中,具有环状结构的C2对于引物多聚体具明显抑制作用,且呈浓度依赖型。结果显示,其抑制效用在终浓度75~150 nmol/L最佳(图1B);而具发卡结构的C1对于引物多聚体均无明显抑制效果(图1C)。
2.2 单链环状DNA C2对DNA聚合酶浓度的影响 以筛选出来的C2的最佳浓度为基准,利用已经成功建立的STAT1的基因扩增体系研究DNA聚合酶浓度与C2的相关性,结果显示,无论是Hot-start DNA聚合酶还是普通DNA聚合酶,在含有C2的PCR体系中,最低终浓度不得少于0.025 U/μl(图1D)。
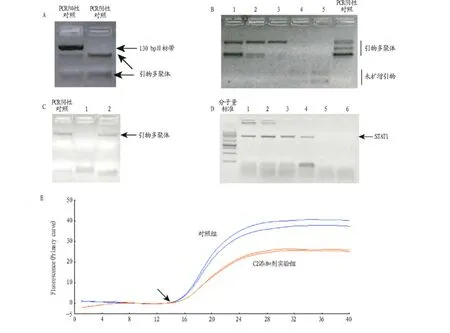
图1 利用单链环状DNA添加物抑制引物多聚体
2.3 单链环状DNA C2对扩增产物产量的影响 利用实时定量PCR来研究单链环状DNA C2是否会影响PCR扩增产物产量,通过比较含有或不含有C2的不同反应体系的Ct平均值,结果显示两者无显著差异(图1E),说明单链环状DNA C2的添加对PCR产物影响不显著(P>0.05)。
3 结论与讨论
由于部分引物多聚体副产物与Ditag目的片段长度非常接近,不仅影响目的产物的产量,同时在经过DNA胶回收及后续处理步骤后,在导致测得的序列中将获得大量的未知序列,有可能被误判读成新SAGE tag。引物多聚体的出现显示,作为SAGE技术中最关键的环节,Ditag接头序列和PCR引物在设计上存在很大的缺陷。
试验设计的单链DNA添加物C1和C2主要序列和结构上非常相似,只是C2是一个封闭的单链环状结构,而C1呈一端开放性的发卡结构。但是试验显示,发卡结构的单链DNA添加物C1无法抑制Ditag PCR中引物多聚体的产生;而C2对于引物多聚体的抑制呈现浓度依赖性的特点(图1B、C)。上述结果对于多种普通或热启动DNA聚合酶具有一致性(数据未附)。虽然C2添加剂可以抑制引物副产物的产生,但是在试验筛选出来的浓度范围内,C2不会降低特异性产物的产量,这个结论利用定量PCR试验给予了进一步证实。单链环状DNA添加物C2不仅可以用于SAGE试验,其实对于常规的PCR试验中引物副产物的抑制也有作用,但是其最佳使用终浓度需要进一步筛选和优化。
目前,用于PCR反应的DNA聚合酶种类和来源有很大不同,但是在结构具有相似性[8-11]。试验认为引物多聚体的产生是伴随PCR试验、却同时独立于其他目的片段扩增的过程,仅与引物和聚合酶有关。因此,从仅含有DNA聚合酶和引物的阴性对照试验就可以发现,这也为研究引物多聚体的产生提供了一个优良的体系。试验虽然发现了C2添加物抑制PCR引物多聚体的现象,但是对其机理还是不甚清楚。一个可能的解释是,C2能够优先有效地竞争性占据DNA聚合酶上对于引物多聚体形成具有关键性的功能域,从而抑制引物多聚体的形成。其具体分子机理尚需要进一步研究。
[1]KLOPFLEISCH R,GRUBER A D.Transcriptome and Proteome Research in Veterinary Science:What Is Possible andWhat Questions Can Be Asked?[J].The Scientific World Journal,2012,2012:25496 -254975.
[2]SCHUSTER S C.Next-generation sequencing transforms today’s biology[J].NatMethods,2008,5(1):16 -8.
[3]李靖,孔祥银.基因表达系列分析技术的新进展[J].生物工程学报,2001,17(6):613 -616.
[4]VELCULESCU V E,ZHANG L,VOGELSTEIN B,et al.Serial analysis of gene expression[J].Science,1995,270(5235):484 -487.
[5]HORANM P.Application of serial analysisof geneexpression to the study of human genetic disease[J].Hum Genet,2009,126(5):605 -614.
[6]PATINOW D,MIAN O Y,HWANG PM.Serial analysis of gene expression:technical considerations and applications to cardiovascular biology[J].Circ Res,2002,91(7):565 -569.
[7]BROWNIE J,SHAWCROSS1 S,THEAKER J,et al.The elimination of primer-dimer accumulation in PCR[J].Nucleic Acids Research,1997,25(16):3235-3241.
[8]STEITZ T A.DNA Polymerases:Structural diversity and common mechanisms[J].JBiol Chem,1999,274(25):17395 -17398.
[9]BEESE L S,DERBYSHIRE V,STEITZ T A.Structure of DNA polymerase IKlenow fragment bound to duplex DNA[J].Science,1993,260(5106):352-355.
[10]EOM S,WANG J,STEITZ T A.Structure of Taq polymerasewith DNA at the polymerase active site[J].Nature,1996,382(6588):278 -281.
[11]LIY,MITAXOV V,WAKSMAN G.Structure-based design of Taq DNA polymeraseswith improved properties of dideoxynucleotide incorporation[J].Proc Natl Acad Sci USA,1999,96(17):9491 -9496.
Using Single Strand Circular DNA Additive to Inhibit Primer PolymersGenerated during Ditag PCR of Serial Analysisof Gene Expression
LIU Yu-hui,ZHENG Dong et al (Northeast Forestry University,Harbin,Heilongjiang 150040)
[Objective]To investigate the hypothesis thatusing single strand DNA asadditive to preventmispriming productof PCR.[Method]We designed and synthesized two kinds of single strand DNA additives;one was circular and another hairpin-like.Different concentrations of each kind of DNA additiveswere added into Ditag PCR systems of Long Serial Analysis of Gene Expression.Final productswere used for gel electrophoresis to screen DNA additives structure-or concentration-dependent inhibition of PCR primer polymer.[Results]Single strand circular buthairpin-like DNA additiveswas able to inhibit PCR primer polymer;the optimal final concentration was between 70 and 150 nmol/L,and single strand circular DNA additiveswill notaffect differential expression of SAGE tags.[Conclusion]Single strand circular DNA can be used as effective inhibitor to prevent primer polymers in PCR amplification.
Single strand circular DNA(sscDNA);Serial analysis of gene expression(SAGE);Primer polymers;Inhibition
S188
A
0517-6611(2014)15-04576-03
黑龙江省博士后科研启动资助金(项目编号:LBH-Q07009);中央高校基本科研业务费专项资金(项目编号:2572014EA05)。
刘玉慧(1989-),女,黑龙江哈尔滨人,硕士研究生,研究方向:分子细胞生物学。*通讯作者,教授,从事分子细胞生物学研究。
2014-04-29
猜你喜欢
现代实用医学(2022年10期)2022-12-08
中国民间疗法(2021年9期)2021-07-22
中国海洋大学学报(自然科学版)(2019年2期)2019-12-07
天然产物研究与开发(2018年10期)2018-11-06
现代检验医学杂志(2016年4期)2016-11-15
现代检验医学杂志(2016年5期)2016-08-20
上海农业学报(2016年5期)2016-02-10
解放军医学院学报(2015年10期)2015-03-21
中国医科大学学报(2015年10期)2015-03-01
组合机床与自动化加工技术(2014年12期)2014-03-01
