
具抗癌活性的菲并吲哚里西丁类生物碱构效关系及不对称全合成研究进展
2014-01-09 05:07:54祁利民
天然产物研究与开发 2014年11期
杨 晋,祁利民
1北方民族大学化学与化学工程学院;2 国家民委化工技术重点开放实验室,银川 750021
生物来源的菲并吲哚里西丁类生物碱主要分布在萝藦科的娃儿藤属(Tylaphora)、鹅绒藤属(Cynanchum)和桑科的榕属(Ficus),此外,在班蝶科的班蝶属(Ideopsis)中也发现有该类生物碱的存在。自1935年首次从印度药用植物印度娃儿藤(Tylaphora indica)中发现娃儿藤碱(tylophorine,1)以来,已分离得到该类化合物近80 个[1,2]。这类生物碱具有抗病毒、抗炎等多种生物活性,其中,最引人注目的是抗肿瘤活性,根据现有文献报道,已发现的化合物中约25%具有抑制肿瘤细胞生长的活性,引起了药学家和化学家的极大兴趣,相继展开了该类生物碱的全合成工作。但是,这类化合物在拥有抗癌活性的同时,也有一定的毒副作用,例如密花娃儿藤碱(tylocrebrine)在一期临床研究中发现具有很高的中枢神经作用而终止研究[3],从而制约了这类高活性生物碱的进一步开发利用。因此,以菲并吲哚里西丁类生物碱为先导化合物,明确构效关系,利用现代化学和生物学的方法,在不影响抗肿瘤生物活性的前提下,降低毒性,是解决这类生物碱开发的必由之路。本文对具有抗肿瘤活性的菲并吲哚里西丁类生物碱的构效关系和不对称全合成研究进展进行了综述。
1 构效关系
已报道的全合成和结构修饰工作为菲并吲哚里西丁类生物碱的构效关系提供了大量可供分析的数据,这些数据提示我们,菲并吲哚里西丁类生物碱化学结构中菲环的取代基、C14位是否被氧化、N 原子的存在形式都可能影响这类生物碱的抗肿瘤活性。
1.1 菲环
天然存在的菲环开裂的菲并吲哚里西丁类化合物,如secoantofine(5)等,通常表现出较弱的抗肿瘤活性[4-6](见表1 和图1),但是,tyloindicine I(12)是一个例外,在美国国家癌症研究中心的抗肿瘤药物筛选中表现优异,是最有可能开发为药物的先导化合物之一[7]。这个化合物的化学结构中包含了一个高度不饱和的吲哚环,可能是其具有高抗肿瘤活性的原因之一。因此,对于菲并吲哚里西丁类生物碱,就抗肿瘤活性来说,拥有较长的共轭链,并具有两个芳香环,是必要的。
表1 所列具有抗肿瘤活性的化合物,在C2上均连有甲氧基,说明C2上连有的基团对该类化合物的抗癌活性影响显著[4,8]。当C6上甲氧基被羟基取代后,抗肿瘤活性进一步增强,如化合物3 对肿瘤细胞的生长抑制作用要强于娃儿藤碱[4,9];如果2,3-二氧甲基被2,3-二氧亚甲基取代,则活性大大降低[9]。Kim 小组[10]对安妥分(antofine,2)菲环上不同取代基对抗癌活性的影响进行了研究,在C2连有甲氧基的右旋安妥分对HCT116 肿瘤细胞株抑制作用的IC50值为9.9 nM,如果在C2上引入异丙氧基或与C3位形成亚甲二氧基,抗肿瘤活性将降低100倍。另外,在保证C2位上为甲氧基的同时,在C6上引入羟基,也会增强化合物抑制肿瘤细胞生长的活性。这一结果提示,具抗癌活性的菲并吲哚里西丁类生物碱的C2上不能连有较大空间位阻的基团,而且在与受体结合时,如果C6上的羟基可能与受体形成了氢键。
1.2 N 原子
天然存在的菲并吲哚里西丁类生物碱中吲哚环上的N 原子存在形式有两种:游离N 原子和氮氧化物。通常,形成氮氧化物的化合物对于肿瘤细胞的生长抑制作用要比相应的化合物低5~10 倍[4,11]。有研究表明,肿瘤细胞生长环境的pH 值与该类生物碱的pKa 越接近,则该类生物碱的细胞抑制活性越显著,如娃儿藤碱在pH 7.0 环境下的细胞毒活性要高于在pH 5.8 环境下呈现的活性,这一现象提示,非质子化的N 原子可能更有利于与受体结合从而发挥抑制作用[12]。
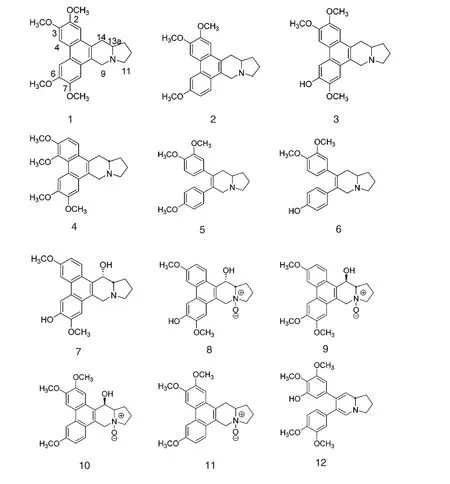
图1 部分具有抗肿瘤活性的菲并吲哚里西丁类生物碱的化学结构Fig.1 Chemical structures of phenanthroindolizidine-type alkaloids with antitumor activity

表1 部分菲并吲哚里西丁类生物碱的抗癌活性Table 1 The antitumor activity of some phenanthroindolizidine-type alkaloids
1.3 立体化学
右旋安妥分较消旋体的抗肿瘤活性高2 倍有余,说明消旋体中可能只有一种对映体具有活性[10]。天然存在的菲并吲哚里西丁类生物碱C13a通常为S 构型,将它们的抗肿瘤活性与合成的、具有C13a(R)构型的化合物相比,可以发现一个有趣的现象:天然的、具有C13a(S)构型的生物碱对HCT116和A549 肿瘤细胞株表现出较强的抑制作用,而合成的、具有C13a(R)构型的生物碱对KB 肿瘤细胞株则抑制作用较强[4,10,13,14]。在C13a上引入羟基等基团,可以有效地增加化合物的水溶性[15],有利于新药开发,因此,由C13a的立体化学带来的构效关系还有待进一步的研究确认。
部分天然的菲并吲哚里西丁类生物碱在C14上存在羟基,如娃儿藤定碱(tylophorindine,7)。有研究报道了娃儿藤定碱和娃儿藤碱等类似物对KB、HepG2 等肿瘤细胞株的抑制作用,结果提示,具有C14羟基的生物碱,C14的立体构型是S 构型的活性要优于R 构型[5,13,16],这与天然发现的具C14羟基的菲并吲哚里西丁类生物碱C14大多为S 构型相一致。
2 菲并吲哚里西丁类生物碱的不对称全合成途径
菲并吲哚里西丁类生物碱的全合成工作始于1950年代,随着外消旋的小穗苎麻素和娃儿藤碱的合成并证明其具有与天然来源的生物碱相似的生物活性,有机化学家们开始研究全合成这类生物碱,娃儿藤碱和安妥分是最常使用的目标化合物,这两个化合物的分子结构中都存在有手性中心。如前所述,菲并吲哚里西丁类生物碱的立体化学影响着其抗肿瘤活性,能够合成非对映异构体,不论是开展构效关系研究,还是开发新药,都是非常理想的目标。因此,手性合成是本文综述的重点。
2.1 通过手性底物进行不对称合成
1983年,Rapoport 小组完成了(S)-(+)-娃儿藤碱的全合成。在合成中,首先利用藜芦醛(veratraldehyde)和3,4-二甲氧基苯乙腈通过羟醛缩合、氧化偶联关环构建了菲环单元,然后再与(S)-(+)-二异丙基谷氨酸酯作用得到胺12,胺12 通过成环、水解得到酸13,酸13 可以非常容易地发生分子内的Friedel-Crafts 反应得到具有绝对构型的酮14,酮14 在三仲丁基硼氢化锂的催化下将14 位的羰基还原为羟基,并进而在氢氧化铅和碳的催化加氢及四氢铝锂还原可以得到(S)-(+)-娃儿藤碱;同时,还可以得到两个在C14位上具有羟基的对映异构体15、16(图2)[17],这些化合物都具有良好的抗肿瘤活性。虽然Rapoport 小组得到的(S)-(+)-娃儿藤碱与天然来源的娃儿藤碱是对映异构体,但是通过引入(S)-谷氨酸衍生物作为手性试剂,在成功合成具有绝对构型的菲并吲哚里西丁生物碱的同时,确定了天然来源娃儿藤碱的绝对构型。

图2 路线1Fig.2 Scheme 1
Nordlangder 和Njgoroge 报道了一条相对较短的(S)-(+)-娃儿藤碱的合成路线。在这条全合成途径中所使用的手性底物是(S)-N-(三氟乙酰)-L-脯氨酰氯,手性底物与2,3,6,7-四甲基菲发生Friedel-Craft 反应生成(S)-N-(三氟乙酰)-2,3,6,7-四甲基-9-L-脯氨酰基菲,然后经还原羰基、脱去三氟乙酰基、关环得到(S)-(+)-娃儿藤碱(图3)[18,19]。

图3 路线2Fig.3 Scheme 2
Furstner 小组利用D-(-)-吡咯烷为手性底物,通过延伸侧链的方法首先构建了吲哚环的片段,含有炔基锂的吲哚环片段与多取代二苯发生Kumada偶联,经Pictet-Spengler 反应,合成了(R)-(-)-安妥分(图4)[20]。值得注意的是,利用这条合成路线合成的安妥分的立体化学与天然得到的安妥分一致。
Kim 小组利用(R)-1-三丁基锡基-1-丁烯-3-醇为手性底物,与菲基溴反应,生成的具有手性的丙烯醇与三氯乙腈发生Pinner 反应生成亚胺基醚,该化合物脱去三氯乙酰保护基团,经Pictet-Spengler 反应,合成了(R)-(-)-安妥分(图5)[21]。

图4 路线3Fig.4 Scheme 3
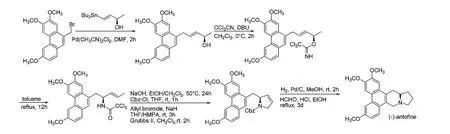
图5 路线4Fig.5 Scheme 4
我国学者在娃儿藤碱的全合成方面也做了大量的工作。Huang 小组的合成路线使用的手性底物是叔丁基L-焦谷氨酸酯,手性底物与菲基氯发生Friedel-Craft 反应生成(S)-(+)-娃儿藤碱(图6)[22]。这条合成路线得到的产物具有很高的光学纯度(92%,=+74.9°)。

图6 路线5Fig.6 Scheme 5
2.2 利用手性催化剂进行不对称合成
2003年,Kim 小组在完成安妥分的全合成研究时,使用了相转移催化剂,该催化剂是一个手性催化剂17,使菲基溴与具亚胺结构的甘氨酸衍生物反应时,得到的亚氨基醚18 具有97% 光学纯度,经Pictet-Spengler 环合反应,即可得到(R)-(-)-安妥分(图7)[23]。

图7 路线6Fig.7 Scheme 6

图8 路线7Fig.8 Scheme 7
Chemler 小组采用了不同的构建菲环的合成途径。以多取代的苯乙醇为原料,使之生成溴代产物,然后二聚生成二苯乙烷,在phenyliodine(Ⅲ)bis(trifluoroactetate)(PIFA)的作用下环合成菲环,再与pentenylamine 生成中间产物19,在环合时,采用了含有二价铜离子的手性催化剂20,在脱去二氧化硫基团后,再环合即得到(S)-(+)-娃儿藤碱(图8)[24]。
2.3 使用手性试剂进行不对称合成
Fukumoto 小组将多甲氧基取代的二苯乙烯酸与4-氨基丁先二乙基缩醛反应,生成化合物21,该化合物在具有手性的(-)苄基薄荷基三苯膦醋酸酯[(-)-phenylmentyl (triphenylphosphoranylidene)acetate]作用下发生Horner-Madsworth-Emmons 反应生成化合物22,化合物在TBSOTf 和三乙胺作用下发生双Michael 重排得到与天然娃儿藤碱旋光一致的手性前体,再经还原,即可得到(-)-娃儿藤碱(图9)[25]。
2.4 使用手性辅助剂进行不对称合成

图9 路线8Fig.9 Scheme 8

图10 路线9Fig.10 Scheme 9
Commins 小组采用手性辅助剂(-)trans-2-(αcumyl)cyclohexyl ester 完成了(-)-娃儿藤碱的全合成。首先,吡啶盐在手性辅助剂的引导下与格式试剂3-丁烯基溴化镁发生非对映选择性的加成反应,生成的化合物23 具有很高的光学纯度;该化合物经脱去手性辅助剂后即可得到单不饱和的吲哚环,不饱和的吲哚环催化加氢,与3,4-二甲氧基苯基溴化锌反应生成(-)-septicine,这个化合物也存在于天然界;(-)-septicine 在VOF3作用下即可生成(-)-娃儿藤碱(图10)[26]。
3 结语
菲并吲哚里西丁类生物碱是一类具有较高活性的天然抗肿瘤生物碱,一直是药物化学家们研究的热点。虽然有大量的研究报告,但是依旧还有未能阐明的构效关系,如13、14 位的光学特性是否对抗肿瘤活性有影响,还更多的数据来验证。
同时,菲并吲哚里西丁类生物碱的全合成也是研究热点之一。对于这类化合物的全合成策略,可以明显地分为两类:先构建菲环和先构建吲哚环。先构建菲环,再进行碳链延伸,与五元含氮杂环连接,然后关环形成目标化合物的合成策略是目前合成这类化合物的主流。这种合成策略通常合成步骤在10 步以上,较为繁琐。采用含氮杂环先构建吲哚环,然后与多取代苯连接,再关环形成目标化合物的全合成路线,相关报道较少,而且反应中可能会使用到一些较为苛刻的条件,实用性略显不足。根据菲并吲哚里西丁类生物碱的分子结构特点,发展更为简便、经济的合成路线,或者在了解构效关系的基础上,进行结构改造或结构修饰,是更好地利用和开发这类生物碱的有效途径。
1 Li Z,et al.Isolation,Total synthesis and biological activity of phenanthroindolizdine and phenanthroquionlizidine alkaloids.Synthesis,2001,16:2365-2378.
2 Zhang C(张成刚),Tan XD(谭显东).Advances in phenanthroindolizine alkaloids research.J Sichuan Normal Univ,Nat Sci(四川师范大学学报),2005,28:366-370.
3 Suffness M,Douros J.Anticancer agents based on natural product models.Beijing:Academic Press,1980,465-487.
4 Staerk D,et al.In vitro cytotoxic activity of phenanthroindolizedine alkaloids from Cynanchum vincetoxicum and Tylophora tanakae against drug-sensitive and multidrug-resistant cancer cells.J Nat Prod,2002,65:1299-1302.
5 Gao W,et al.Structure-activity studies of phenanthroindolizedine alkaloids as potential antitumor agents.Bioorg Med Chem Lett,2007,17:4338-4342.
6 Steark D,et al.Cytotoxic activity of some phenanthroindolizedine N-oxide alkaloids from Cynanchum vincetoxicum.J Nat Prod.,2000,63:1584-1586.
7 Chemler RS.Phenanthroindolizidines and phenanthroquinolizidines:promising alkaloids for anti-cancer therapy.Curr Bio Comp,2009,5:2-19.
8 Su C,et al.Total synthesis of phenanthroindolizidine alkaloids (±)-antofine,(±)deoxypergularinine,and their dehydrocongeners and evaluation of their cytotoxic activity.Bioorg Med Chem,2008,16:6233-6241.
9 Toribio A,et al.Novel seco-dibenzopyrrocoline alkaloid from Cryptocarya oubatchenisis.Org Lett,2006,8:3825-3828.
10 Fu Y,et al.Synthesis and structure-activity studies of antofine analogues as potential anticancer agents.Bioorg Med Chem Lett,2007,17:97-100.
11 Damu AG,et al.Cytotoxic phenanthroindolizidine alkaloids from the roots of Ficus septica.Planta Med,2009,75:1152-1156.
12 Grant P,Sanchez L,Jimenez A.Cryptopleurine resistance genetic locus for a 40s ribosomal component in Saccharomyces cerevistice.J Bacieriology,1974,120:1308-1314.
13 Gao W,et al.Novel mode of action of tylophorine analogs as antitumor compounds.Cancer Res,2004,64:678-688.
14 Gao W,et al.Structural analogs of tylophora alkaloids may not be functional analogs.Bioorg Med Chem Lett,2008,18:704-709.
15 Kwon Y,et al.Design,synthesis,and evaluation of a watersoluble antofine analogue with high antiproliferative and antitumor activity.Bioorg Med Chem,2013,21:1006-1017.
16 Huang X,et al.Cytotoxic alkaloids from the roots of Tylophora atrofolliculata.Planta Med,2004,70:441-445.
17 Buckley TF,Papoport H.α-Amino acids as chiral educts for asymmetric products:chirally specific synthesis of tylophorine and cryptopleurine.J Org Chem,1983,48:4222-4232.
18 Moremo L,et al.A new entrance to the preparation of phenanthrene and phenanthrenooid heterocycles.Synthesis,2001,17:1161-1163.
19 Moremo L,et al.A simple route to new phenanthro-and phenanthroid-fused thiazoles by a PIFA-mediated (hetero)biaryl coupling reaction.Eur J Org Chem,2002,2126-2135.
20 Furstner A,Kennedy JWJ.Total synthesis of the tylophora alkaloids cryptopleurine,(-)-antofine,(-)-tylophorine and(-)-ficuseptine C.Chem Eur J,2006,12:7398-7410.
21 Kim S,et al.Asymmetric total synthesis of (-)-antofine and(-)-cryptopleurine using (R)-(E)-4-(tributylstannyl)but-3-en-2-ol.J Org Chem,2004,69:3144-3149.
22 Jin Z,et al.A concise total synthesis of S-(+)-tylophorine.Chin Chem Lett,2004,15:1164-1166.
23 Kim S,et al.First asymmetric total synthesis of (-)-antofine by using an enantioselective catalytic phase transfer alkylation.Org Lett,2003,5:2703-2706.
24 Zeng W,Chemler SR.Total synthesis of (S)-(+)-tylophorine via enantioselective intramolecular alkene carboamination.J Org Chem,2008,73:6045-6047.
25 Ihara M,et al.Asymmetric total synthesis of naturally occur-ring (R)-(-)-enantiomer of tylophorine via intramolecular double Michael reaction.J Chem Soc Perkin Trans I,1990:2287-2292.
26 Comins DL,et al.Enantiopure N-acyldihydropyridones as synthetic intermediates:asymmetric synthesis of (-)-septicine and (-)-tylophorine.J Org Chem,1997,62:7435-7438.
猜你喜欢
分子催化(2022年1期)2022-11-02 07:10:30
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
名家名作(2021年7期)2021-08-04 08:16:40
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
数学大王·低年级(2016年9期)2016-05-14 10:20:08
语文学刊(2015年15期)2015-08-15 00:50:27
