
大肠杆菌BL21(DE3)中定向进化突变体库构建的方法比较
2013-12-23 03:50陈菲云张大伟刘森
生物技术世界 2013年10期
陈菲云 张大伟 刘森
(1.三峡大学医学院/分子生物学研究所 湖北宜昌 443002;2.中国科学院天津工业生物技术研究所 天津 300308)
对三维结构及其功能之间的相互关系并不明确的酶进行改造,定向进化方法是很好的选择。定向进化就是利用自然选择的方法使工程蛋白能够获得天然蛋白不具备的特定性质的一种方法[1]。它不需要对蛋白结构与功能的关系有深刻的了解,它仅通过随机突变或基因重组技术建立起突变体文库,然后连接到表达载体导入宿主内进行蛋白表达,具有改进性状的突变体被筛选出来再进行下一轮突变直至得到符合要求的突变体。文献资料已有大量关于利用定向进化成功改变了分子的特异性和催化性能的例子[2]。
定向进化的步骤包括DNA突变库的体外构建和DNA突变库向宿主细胞的转化。其中DNA突变库的体外构建的常见方法已有不少文章总结过[3,4],本文选择的是简单高效的易错PCR法。DNA突变库向宿主细胞的转化是至关重要的一步,因为库容量的大小受转化效率的限制,从而影响获得好的突变体的机率。然而应用广泛的表达宿主菌大肠杆菌虽背景清楚,生长繁殖快,易操作[5],是最常用的定向进化宿主菌。但其中作为蛋白表达常用的BL21(DE3)的转化效率仅是DH5α的十分之一,不利于突变体库的建立。综上,高效的克隆转化方法是定向进化方法所需求的。
做克隆最常用的是酶切连接法,是分子实验常规的一种分子操作方法。但是在实际操作过程中,酶切连接法有一定的局限性,存在操作繁琐,耗时长,且转化率低,不适于建库等需要大容量克隆的实验。其它的克隆转化的方法也已经有很多种[6,7,8,9,10],我们选取了其中简便高效的PCR法直接获得多聚质粒的方法做突变体库,与已有的常规方法比较。
通过比较两种克隆转化方法,分析两种方法的应用效果。从而选择高效的克隆转化方法作为突变体库构建方法,为下一步高通量筛选奠定良好的基础,也为其他人做定向进化突变体库提供了很好的参考。
1 材料与方法
1.1 材料
质粒pET21a-lysozyme是本实验室构建,pET21a载体为本实验室自存。大肠杆菌BL21(DE3)感受态细胞购自北京全式金生物技术有限公司。内切酶,T4连接酶及Q5超保真DNA聚合酶均为New England Biolabs公司产品。Agorose是Biowest的Regular agarose G-10。核酸电泳系统是北京六一仪器厂产品。Pcr仪购自东胜国际贸易有限公司。易错pcr试剂盒购自北京天恩泽基因科技有限公司。
1.2 常用的克隆转化方法构建突变体库
1.2.1 基于pcr的随机突变
以质粒pET21a-lysozyme为模板,用易错pcr法构建lysozyme序列的随机突变库,首先根据溶菌酶序列设计两条引物,引物为表1中的Lysozyme F和Lysozyme R。为了得到突变体库,易错PCR的反应体系中添加了Mn2+离子,而且反应所用的四种dNTP不是等比例的,我们用的是即用型易错PCR试剂盒,其各项试剂的比例是经过优化的。
50uL易错pcr体系如下:5uL 10×易错pcr缓冲液,5uL 10×易错pcr专用dNTP,5uL MnCl2(5mM),1uL模板质粒pET21alysozyme(10 ng/uL),各1uL PCR引物F/R(10 uM),1uL TaqDNA聚合酶(5U/uL),ddH2O补至50 uL。
PCR扩增条件:94℃ 3min;94℃ 1min,60℃ 1min,72℃ 1 min,30个循环;72℃ 10min。
易错pcr扩增产物即为lysozyme序列的随机突变库。
1.2.2 突变文库的构建
PCR产物经琼脂糖凝胶分离,胶回收纯化后,限制性内切酶NdeI和XhoI分别对易错PCR扩增产物和质粒pET21a-lysozyme进行酶切。酶切后的PCR产物纯化,酶切后的载体经琼脂糖凝胶分离,切胶回收线性化载体片段,然后将酶切纯化后的PCR产物和酶切胶回收的载体按照物质的量3:1以T4连接酶连接体系进行连接。
将连接产物转化至大肠杆菌BL21(DE3)化学感受态细胞或将连接产物纯化后,转入大肠杆菌BL21(DE3)电转化感受态细胞。涂布于LB(含100ug/uL的Amp)平板。37℃恒温培养12h后,察看单菌落数量。
1.3 PCR产物直接转化方法构建突变体库
(1)此方法的引物设计及流程(图1),首先仍然是以质粒pET21alysozyme为模板,用易错pcr法构建lysozyme序列的随机突变库,引物为Table 1的P1,P2,去掉了酶切位点两边的保护碱基,以便于Overlap PCR的顺利进行。

表1 文中所用引物汇总 (Table 1 Oligonucleotides used in this study.)
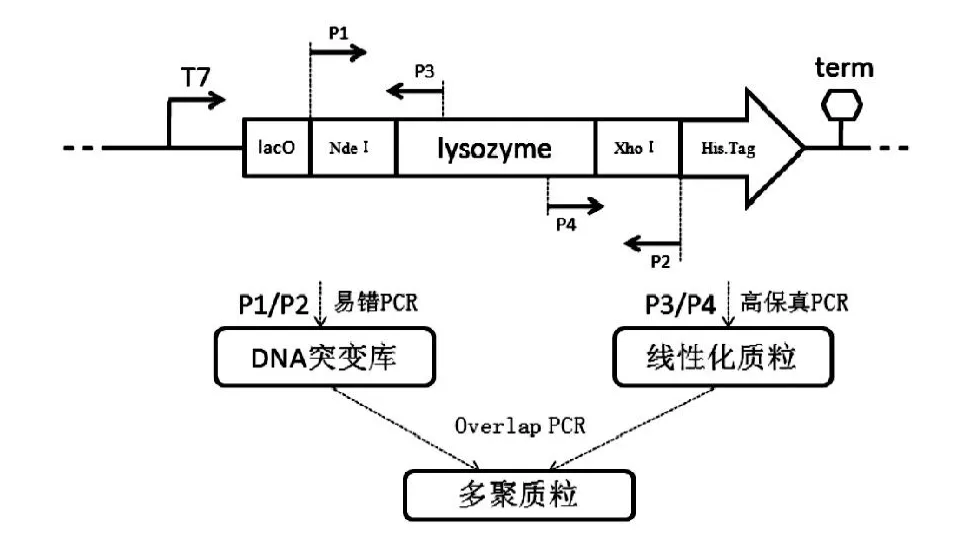
图1 新方法构建突变体库实验流程P1,P2是易错PCR引物;P3,P4是质粒线性化引物;lysozyme是我们突变体库的目的序列,其它是质粒上的一些基本元件。Fig 1 The flow scheme of the two-step PCR procedure for the gene mutagenesis and plasmid multimerization.Lysozyme is the purpose sequence of our mutant library,others are the basic components of the plasmid.P1,P2,P3 and P4 denote the positions of the primers for the PCR amplication.
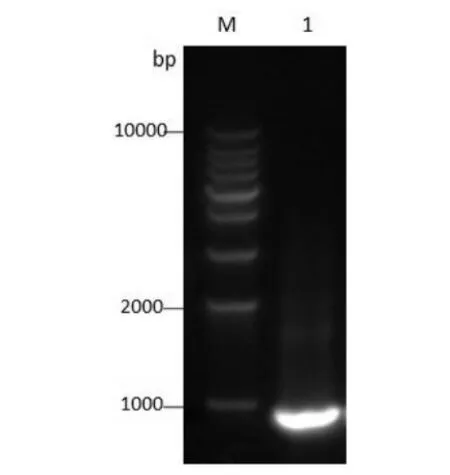
图2 常规方法建立突变体库过程中易错PCR产物的琼脂糖凝胶电泳图M是marker;1是易错PCR产物Fig.2 Error-prone PCR products of the routine method.M:1Kb DNA marker;1:error-prone PCR products

图3 两种克隆转化方法所得克隆数量比较1是常规方法做克隆转化所得克隆数量,2是新方法所得的克隆数量Fig.3 The number of clones obtained by the two methods in Constructing Mutant Library in E.coli BL21(DE3).1:Result of transformation by routine method 2:Result of transformation by new method.
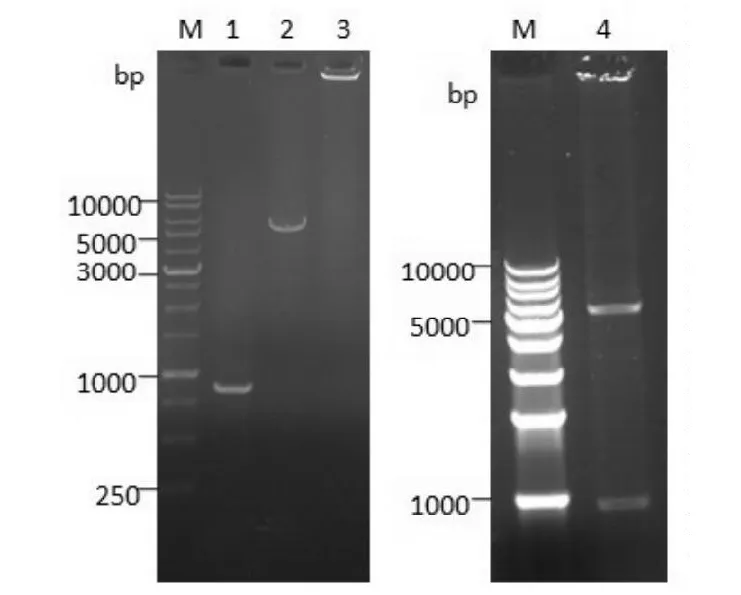
图4 多聚PCR法直接转化构建突变体库的凝胶电泳图M是marker;1是易错PCR产物;2是线性化质粒PCR产物;3是overlap PCR 产物多聚化质粒;4是多聚质粒用NdeI和XhoI酶切后的产物Fig.4 Analysis of a 1%agarose gel for the PCR products of New method.M:marker;1:Error-prone PCR products of new method;2:PCR linearized vector;3:DNAmultimer generated by overlap extension PCR;4:Multimer digested with two restriction enzymes
PCR体系及扩增条件同1.2.1。
(2)通过PCR将质粒pET21a-lysozyme线性化。通过高保真PCR线性化质粒pET21a-lysozyme,引物设计时,要使线性化质粒包含要突变的溶菌酶的一部分序列,以便于下一步overlapPCR线性化质粒与易错PCR序列有重叠区,引物为表1中P3,P4。
50uLPCR反应体系如下:10uL 5×buffer,5uL 10×dNTP Mix,1uL模板质粒pET21a-lysozyme(10ng/uL),各1uL PCR引物F/R(10uM),0.5uL Q5超保真DNA聚合酶(2000U/uL),ddH2O补至50uL。
PCR扩增条件:98℃ 2min;98℃ 15 s,55℃ 30 s,72℃ 3min,30个循环;72℃ 10min。
(3)通过Overlap PCR得到多聚化的重组质粒。线性化后的质粒和易错pcr的产物均通过琼脂糖凝胶电泳分离检测,然后用凝胶回收试剂盒回收所需条带,测定浓度。
Overlap PCR的反应体系:10uL 5×buffer,5uL 10×dNTP Mix易错pcr产物(体系终浓度2ng/uL),线性化质粒(与易错pcr产物等摩尔质量),0。5uL Q5超保真DNA聚合酶(2000U/uL),ddH2O补至50uL。
扩增条件:98℃ 3 min;98℃ 15 s,55℃ 30s,72℃ 5min,30个循环;72℃ 10min。
(4)产物鉴定。将Overlap PCR得到的多聚化的重组质粒用醇沉法纯化后,用限制性内切酶NdeI和XhoI酶切,以鉴定所得PCR产物是否是含有溶菌酶序列的pET21a质粒。
将通过Overlap PCR得到的多聚化的重组质粒10uL转化至大肠杆菌BL21(DE3)化学感受态细胞,涂布于LB(含100ug/uL的Amp)平板。37℃恒温培养12h后,察看单菌落数量。(表1)
2 结果
2.1 常规方法构建突变体库
首先构建了重组质粒pET21a-lysozyme,重组质粒包含一段954bp的溶菌酶基因,以质粒pET21a-lysozyme为模板,在溶菌酶基因两端设计引物以易错pcr法得到lysozyme序列的随机突变库。经琼脂糖凝胶分离鉴定:在marker 1000bp靠下的位置得到一条明亮单一的条带(图2),由此可见易错PCR产物为所需目的片段。
质粒酶切后经琼脂糖凝胶分离,将线性化载体片段切胶回收与PCR酶切后的片段连接后转入大肠杆菌BL21(DE3)感受态细胞。用此种方法做突变库,无论是用化学转化法还是转化效率相对较高的电转化法,得到的单菌落数量均不多,一再提高之前做连接所用的质粒线性片段与PCR酶切产物的量也不能解决问题。每个平板所得的单菌落数量始终在50以下(图3中1)。
2.2 多聚PCR产物直接转化法构建突变体库
此方法仍然从质粒pET21a-lysozyme出发,经琼脂糖凝胶鉴定仍可见明亮的与目的产物大小相符的条带(图4的1)。第二步引物设计在溶菌酶基因内部,这样线性化质粒与溶菌酶基因两边均有100bp重叠序列。于是,线性化质粒的全长序列为5620bp,PCR完成后经琼脂糖凝胶鉴定,与目的片段大小相符(图4的2)。将上两步PCR所得产物经琼脂糖凝胶电泳分离并切胶回收目的片段后,测定所得的两个目的片段浓度,然后按1:1比例做第三步的overlap PCR,此次PCR产物为多聚质粒,分子量过大,产物只是聚集在点样孔内(图4的3)。为了验证所得产物为目的质粒,我们将overlap PCR的产物纯化后用限制性内切酶NdeI和XhoI酶切,酶切产物经琼脂糖凝胶电泳后可见明显的两条带,其大小与我们的质粒pET21a和插入片段大小相符(图4的4)。
将所得的Overlap PCR产物直接以化学转化方法转入大肠杆菌BL21(DE3)感受态细胞,涂布在含有氨苄的固体平板上,长出的单菌落库即为预期随机突变体库。用此种方法建突变体库,效率很高,平均每个平板的单菌落数量均在200以上(图3的2)。而且以此种方法做克隆,每管overlap PCR产物可转化至少5个平板,单次产出高。
3 讨论
在以BL21为宿主表达目的蛋白的过程当中,一般是将目的基因首先用不含有T7 RNA聚合酶基因的宿主菌进行克隆,比如DH5α。完成克隆后,将质粒转入染色体上带有lac UV 5调控的T7 RNA聚合酶基因的表达宿主菌中,用IPTG诱导表达目的蛋白。这样一则是为了避免对宿主细胞有毒的蛋白产物影响质粒的稳定性。一则是由于BL21的转化效率低,仅是DH5α克隆型宿主菌的转化效率的十分之一,不利于常规方法做克隆时连接产物的转化。但是由于我们要建立基因突变库,需要一步到位得到大量单克隆,先转化DH5α再提质粒转化BL21会降低建库效率且增大后期筛选难度。而BL21的转化效率又确实不利于大量突变体库的获得,于是我们需要改进克隆转化方法。
文献中介绍的PCR直接获得多聚体的克隆转化法在BL21中的转化文章中已实验过[10]。按照文献中的实验方法,结合我们的实验需求,设计实验流程如图1所示,DNA突变库的获得仅需要三步PCR。
与常规克隆转化方法相比,新方法步骤减少,操作简单而且省时高效。常规方法构建突变体库至少需要两天时间,而新方法一天就可以完成。常规方法不仅步骤较多,耗时长,而且操作步骤多造成的DNA突变库损失也大,最后转化效率又很低,所以用常规方法做突变体库太过困难,需要大量重复操作。新方法仅是三步PCR反应,省去了酶切再连接的繁琐,不单节省时间而且避免了酶切连接过程中的产物损失,一轮实验就可以得到大量突变子,是一种简单实用且高效的突变体库建立方法。
综上,本文将常规克隆转化方法与新方法建立突变体库进行了比较,结果表明新方法在定向进化突变体库的构建上具有较大优势,这种简单高效实用的突变体库构建方法值得借鉴。
[1]Heselpoth R D,Nelson D C.A New Screening Method for the Directed Evolution of Thermostable Bacteriolytic Enzymes[J].Journal of Visualized Experiments,2012(69):e4216.
[2]Jackel C,Kast P,Hilvert D.Protein design by directed evolution.Biophysics,2008,37:153-173.
[3]张红缨,孔祥铎,张今.蛋白质工程的新策略—酶的体外定向进化.科学通报,1999,44(11):1121-1127.
[4]贾向东,陈德富,陈喜文等.几种定向进化技术的比较及文库构建策略.中国生物工程杂志,2003,23(12):68-72.
[5]Dong X,Tang B,Li J et al.Expression and Purification of Intact and Functional Soybean(Glycine max)Seed Ferritin Complex in Escherichia coli [J].J Microbiol Biotechnol,2008,18(2):299-307.
[6]You C,Zhang X Z,Zhang Y H.Simple cloning via direct transformation of PCR product(DNA multimer)to Escherichia coli and Bacillus subtilis.Applied and Environmental Microbiology,2012,78(5):1593-1595.
[7]Quan J Y,Tian J D.Circular polymerase extension cloning for highthroughput cloning of complex and combinatorial DNA libraries.Nat Protoc,2011,6(2):242-51.
[8]Li M Z,Elledge S J.SLIC:a method for sequence-and ligation-independent cloning.Methods Mol Biol,2012,852:51-59.
[9]Aslanidis C,de Jong P J.Ligation-independent cloning of PCR products(LIC-PCR).Nucleic Acids Res,1990,Oct 25;18(20):6069-6074.
[10]Oh SK,Kim SB,Yeom SI,Lee HA,Choi D.Positive-selection and ligation-independent cloning vectors for large scale in planta expression for plant functional genomics.Mol Cells,2010,Dec;30(6):557-562.
猜你喜欢
中学生数理化·七年级数学人教版(2022年6期)2022-06-05
中学生数理化·八年级物理人教版(2022年4期)2022-04-26
太原理工大学学报(2021年6期)2021-11-25
中学生数理化(高中版.高考数学)(2021年12期)2021-03-08
中学生数理化(高中版.高考数学)(2020年10期)2020-10-27
中等数学(2020年2期)2020-08-24
中国海洋大学学报(自然科学版)(2019年7期)2019-05-21
中国海洋大学学报(自然科学版)(2019年7期)2019-01-04
测控技术(2018年9期)2018-11-25
北京航空航天大学学报(2016年7期)2016-11-16
