
Determination of cefcapene acid by LC-MS and their application to a pharmacokinetic study in healthy Chinese volunteers
2013-12-23 06:15:04HongFeiDunLiDingXioBingLiLinLinHuAiDongWenYeLengZhenLiu
Hong-Fei Dun, Li Ding,*, Xio-Bing Li, Lin-Lin Hu, Ai-Dong Wen,Ye Leng, Zhen Liu
aDepartment of Pharmaceutical Analysis, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
bDepartment of Pharmacy, The First Affiliated Hospital of the Fourth Military Medical University of PLA, Xi'an 710032, China
1. Introduction
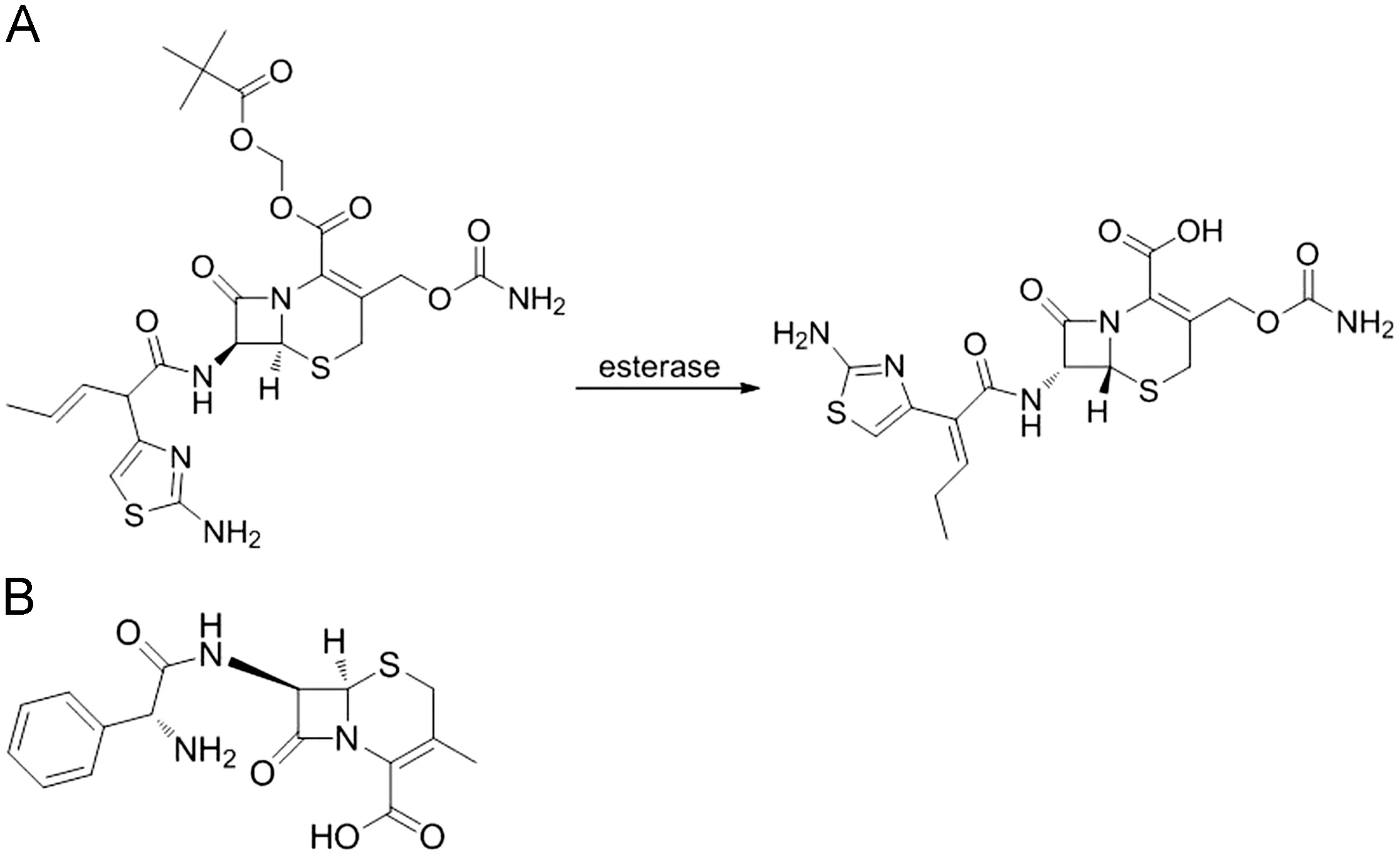
Fig.1 The chemical structures of cefcapene pivoxil and its active metabolite cefcapene acid (A) and the chemical structure of cefalexin (B).
Cefcapene pivoxil (S-1108), an oral cephem antibiotic possessing a pivaloyloxymethyl ester group, is a prodrug of cefcapene acid (S-1006) [1,2]. It is easily deesterified in the intestine in a manner similar to that of other prodrugs having a pivaloyloxymethyl (POM) ester [3-5]. Its active metabolite,cefcapene acid (see Fig.1), is highly active against a wide range of gram-positive and gram-negative bacteria except several bacteria such as Pseudomonas aeruginosa and Enterococci. No publication described the method for the analysis of cefcapene pivoxil in human. By immediately adding potassium fluoride (4 mol/L, 20 μL) and 0.5% folic acid (20 μL) to 200 μL freshly collected plasma, we have solved the problem of instability of cefcapene pivoxil in plasma.Although the method with a lower limit of quantification(LLOQ)of 0.01 μg/mL for the determination of cefcapene pivoxil in human plasma was developed in our laboratory, we still cannot detect cefcapene pivoxil in the freshly collected incurred human plasma. This result implied that the deesterification of cefcapene pivoxil in the intestine was rapid and almost complete. There are a few reports which described the pharmacokinetics of cefcapene acid determined by microbiological assay method [6-9] or isotope labeling method [10].The major drawback of microbiological assay is the lack of specificity. At the same time, isotope labeling cannot be used in human. Nakashima et al. [11] described an HPLC-UV method for the analysis of cefcapene acid in human, in which solid phase extraction must be used in sample preparation. In this paper, we describe two simple, rapid and specific liquid chromatography-mass spectrometry (LC-MS) methods with the LLOQs of 0.03 μg/mL for the determination of cefcapene acid in human plasma and 0.1 μg/mL in human urine, respectively.The fully validated methods have been successfully applied to a pharmacokinetic study of cefcapene acid in health Chinese volunteers receiving single oral doses at 50, 100 and 200 mg of cefcapene pivoxil hydrochloride tablets, as well as multiple oral doses of 100 mg t.i.d. for 7 consecutive days.
2. Experimental
2.1. Chemicals and reagents
The reference standard of cefcapene acid (purity 94.0%, lot no.101101)was supplied by Shandong Luoxin Pharmaceutical group Co. Ltd. (Shandong, China). The internal standard(cefalexin, the IS) was purchased from National Institute for the Control of Pharmaceutical and Biological Products(purity 94.4%, lot no.130408-200710; NICBP, Beijing, China).Methanol of HPLC grade was purchased from Merck KGaA(Darmstadt, German). Ammonium acetate and formic acid were of analytical grade purity and purchased from Nanjing Chemical Reagent Co. Ltd (Nanjing, China). Distilled water was used throughout the study.
2.2. Instrumentation
An Agilent series 1100 LC/MSD VL system was used,consisting of a binary pump, a vacuum degasser, a thermostatic column oven, an autosampler and a single quadrupole mass spectrometer (Agilent Technologies, Palo Alto, CA,USA). An electrospray ionization (ESI) interface in positive ionization mode was used. Data processing was performed with ChemStation software (version 10.02 A).
2.3. Chromatographic and mass spectrometric conditions
A Hedera ODS-2 column, 5 μm, 150 mm×2.1 mm i.d.(Hanbon Science and Technology)with a security Guard-C18(4 mm×2.0 mm,5 μm,Phenomenex)was used for the analyte separation. For the plasma assay, the isocratic mobile phase consisted of 35% solvent A (Methanol) and 65% solvent B(10 mm ammonium acetate buffer solution containing 0.2%folic acid).The flow rate was set at 0.3 mL/min. For the urine assay, the isocratic mobile phase consisted of 30% solvent A(Methanol) and 70% solvent B (10 mM ammonium acetate buffer solution containing 0.2% folic acid). The flow rate was also set at 0.3 mL/min. Chromatography was performed at 35°C using a 6 μL injection volume.
The mass spectrometer equipped with a ESI source was set with a drying gas (N2) flow of 13 L/min, nebulizer pressure of 40 psi, drying gas temperature of 350°C, capillary voltage of 4.0 kV and fragmentor voltage of 125 V. Positive ion mode was employed. Quantitation was carried out in selected ion monitoring (SIM) mode, using target ions at m/z 454.2 for cefcapene acid and m/z 348.2 for the IS.
2.4. Preparation of calibration standards and quality control samples
Primary stock solutions of cefcapene acid (1.0 mg/mL) and cefalexin (1.0 mg/mL) were prepared in methanol. A series of cefcapene acid standard working solutions ranging from 1 to 100 μg/mL were obtained by further dilution of the stock solution with methanol.The working solutions of cefalexin for the internal standard were also prepared by diluting aliquots of stock solution with methanol. All standard solutions were stored at -20°C and were brought to room temperature before use.
Calibration standard and quality control (QC) samples in plasma and urine were prepared by diluting corresponding working solutions with drug-free human plasma and urine,respectively. The concentrations of calibration standard were 0.03,0.1,0.3,0.7,1.5,3,5 μg/mL for plasma and 0.1,0.3,1,3,10, 30, 100, 200, 400 μg/mL for urine, respectively. The concentrations of QC samples were 0.08, 0.6, 4 μg/mL for plasma and 0.2, 20, 350 μg/mL for urine, respectively. All the plasma and urine samples were stored at -80°C.
2.5. Sample preparation
2.5.1. Sample preparation for plasma
Aliquot of 0.2 mL plasma sample was added with 10 μL IS solution (10 μg/mL). After a thorough vortex mixing for 30 s,the mixture was deproteinized with 600 μL methanol and centrifuged at 16,000 r/min for 5 min. Aliquot of 6 μL of the supernatant liquid was injected into the LC-MS system for analysis.
2.5.2. Sample preparation for urine
Aliquot of 0.1 mL urine sample was added with 25 μL IS solution(100 μg/mL).After a thorough vortex mixing for 30 s,the mixture was diluted with 900 μL methanol-water (50:50, v/v), vortexmixed for 30 s,and centrifuged at 16,000 r/min for 3 min.Aliquot of 6 μL of the supernatant liquid was injected into the LC-MS system for analysis.
2.6. Method validation
The methods were validated for specificity,linearity,precision,accuracy, extraction recovery, matrix effect and stability according to the US Food and Drug Administration (FDA)[12] and Chinese State Food and Drug Administration(SFDA)[13]guidance for validation of bioanalytical methods.
2.6.1. Selectivity
The selectivity was assessed by comparing the chromatograms of six different sources of blank biological samples with the corresponding spiked samples. Each blank sample was tested for interferences using the proposed extraction procedure and LC-MS conditions.
2.6.2. Linearity and LLOQ
The linearity was evaluated by assaying calibration curves in biological samples on three consecutive batches. The curves were fitted by a weighted (1/x2) least-squares linear regression method through the measurement of the peak area ratios of the analyte to the IS.The acceptance criterion for a calibration curve was a correlation coefficient(r)of 0.99 or more,and that each back-calculated standard concentration must be within 15% deviation from the nominal value except at the LLOQ,for which the maximum acceptable deviation was set at 20%.
The LLOQ was defined as the lowest concentration in the standard curve at which precision and accuracy should fall within the range of 80-120%. It was established by using five samples independent of standards.
2.6.3. Precision and accuracy
The QC samples were prepared and analyzed on three separate analytical batches to evaluate the accuracy and intra- and inter-day precisions of the analytical method. The accuracy and precisions of the method were determined by analyzing five replicates of the QC samples along with one standard curve on each of three batches.Assay precision was calculated as the relative standard deviation(RSD,%)by use of one-way analysis of variance.The accuracy is the degree of closeness of the determined value to the nominal true value under the prescribed conditions. Accuracy is defined as the relative deviation of the value (E) of a standard from that of its true value (T) expressed as a percentage (RE%). It was calculated using the formula RE%= (E-T)/T×100. Intra- and interday precisions were required to be less than 15%, and the accuracy to be within ±15%.
2.6.4. Extraction recovery
The extraction recovery of cefcapene acid was evaluated by analyzing five replicates at QC levels. The recovery was calculated by comparison of the peak areas of cefcapene acid extracted from biological samples with those obtained from the pure standard without the procedure of extraction.
2.6.5. Matrix effect
The matrix effect is due to co-elution of some endogenous components present in biological samples. These components may not give a signal in SRM of target analytes,but can certainly decrease or increase the response of analytes dramatically to affect the sensitivity,accuracy and precision of the method.Thus assessment of matrix effect constitutes an important and integral part of validation for quantitative LC-MS method for the supporting pharmacokinetic studies [14]. The potential for a matrix effect was evaluated by comparing the peak areas obtained from the analytes added to the reconstituted solutions obtained from blank biological samples, which originated from five different donors and were submitted to the sample-preparation process (A), with those obtained from the analytes dissolved in mobile phase (B). QC samples were evaluated by analyzing five samples at each level. The matrix effect was calculated using the formula matrix effect (%)=A/B×100%. The matrix effect at every concentration level should be less than 15%.
2.6.6. Stability
Stability tests were designed to assess the stability of the analytes under the conditions expected during handling of clinical samples. The stability of cefcapene acid in plasma and urine was studied under a variety of storage and handling conditions by analysis of replicates(n=3)of QC samples.The short-term temperature stability was tested by storing the QC samples at ambient temperature for hours.Freeze-thaw stability(-80°C) was checked through three freeze and thaw cycles.The post-preparative stability of samples in the autosampler was conducted by reanalyzing the extracted QC samples kept under the autosampler conditions (8°C) for hours. The long-term stability was assessed after storage at -80°C for weeks.The stabilities of the stock solution of cefcapene acid and the IS were determined by placing the stock solution at -20°C for 2 months. The results were compared with the freshly prepared solutions.
2.6.7. Diluting study
Five replicate plasma samples (8 μg/mL) were prepared and double diluted with blank plasma. Then the plasma samples were submitted to the sample-preparation process. The percent deviations of accuracy and precision were within±10%.If the concentration levels of plasma samples exceeded the range of standard calibration curve, we would dilute the samples following the diluting study.
2.7. Pharmacokinetic study
2.7.1. Subjects
The established methods were applied in a pharmacokinetic study performed in healthy Chinese subjects. Twelve healthy volunteers(6 males and 6 females, mean age, 34.8±3.2 years; mean weight,60.9±2.7 kg; mean height, 166.6±4.8 cm) were enrolled in this study in Xijing Hospital affiliated to the Fourth Military Medical University, China. The volunteers were free of cardiac, hepatic,renal, pulmonary, neurologic, gastrointestinal and hematologic diseases,as assessed by physical examination,electrocardiography and the laboratory tests including hematology, biochemistry,electrolytes, urinalysis. All the subjects were instructed to abstain from taking any medication for 2 weeks before and during the whole study period.The study protocol was approved by the local Ethical Review Committee in accordance with the principles of the Declaration of Helsinki and the recommendations of the State Food and Drug Administration of China. Written informed consent was obtained from all the subjects.
Each volunteer participated in three phases: phases A, B and C,and there was a 7-day washout period between phases.In phases A and B, 12 volunteers received 50 mg and 200 mg of cefcapene pivoxil hydrochloride tablets at 8:00 am after a standard breakfast, respectively. In phase C, 12 volunteers received 100 mg of cefcapene pivoxil hydrochloride tablet at 8:00 am after a standard breakfast on day 1, and were administered cefcapene pivoxil hydrochloride tablets 100 mg three times a day for 7 days (days 2-8).
In phases A, B and C (day 1), venous blood samples were drawn pre-dose and at 0.25,0.5,0.75,1,1.5,2,2.5,3,4,5,6,8 and 10 h post-dose. In phase C (days 2-8), venous blood samples were drawn before drug administration on days 5, 6 and 7 to determine the Cmin-ss. On day 8, venous blood samples were drawn at 0.25,0.5,0.75,1,1.5,2,2.5,3,4,5,6,8 and 10 h post-dose.
Urine samples were collected in phase C (day 1) in the following time segments:0-2,2-4,4-6,6-8,8-12 and 12-24 h post-dose. All samples were stored at -80°C.
2.7.2. Pharmacokinetic analysis
The data analysis of pharmacokinetic parameters was performed by using Drug and Statistics software (Version. 2.0,Chinese). Non-compartmental pharmacokinetic parameters were calculated for cefcapene acid.The maximum plasma concentration(Cmax) and the time to attain it (Tmax) were noted directly.The area under the plasma concentration-time curve from time zero to the last measurable concentration(AUC0-t)was calculated using the linear trapezoidal rule and was extrapolated to infinity(AUC0-∞) according to the relationship AUC=(AUC0-t+Ct)/Ke. Ctis the last concentration evaluated in plasma which is greater than the lower limit of quantification (LLOQ) and the elimination rate (Ke) was calculated by linear regression of the terminal points of the semi-log plot of plasma concentration against time. The half-life (t1/2) and CL/F were calculated based on the following equations:t1/2=0.693/Ke;CL/F=Ke×Vc.Vcis the apparent volume of distribution of the center compartment,which was estimated by the model after calculation. Vd/F was the apparent volume of distribution, and MRT was mean residence time.
3. Results and discussion
3.1. Conditions for chromatography
Cefalexin was chosen as the internal standard because its chemical properties and mass spectral fragmentation were similar to those of cefcapene acid. When selecting the mobile phase for HPLC-MS system,attention was paid to the influence of mobile phase on the chromatographic retention and the MS sensitivity.Using 10 mM concentration of ammonium acetate buffer, the chromatographic peaks became sharp and symmetrical. Moreover, formic acid could improve the ionization efficiency.Different concentrations of formic acid at levels of 0.05, 0.1,0.2 and 0.3% were tested in the aqueous portion of the mobile phase. The results showed that adding 0.2% formic acid in the aqueous portion was sufficient to achieve the highest MS sensitivity for cefcapene acid and the IS. Finally, acceptable retention time and separation of cefcapene acid were obtained using the following mobile phase: for plasma, an elution system of methanol-10 mM ammonium acetate buffer containing 0.2%formic acid(35:65,v/v)with a flow rate of 0.3 mL/min;for urine,an elution system of methanol-10 mM ammonium acetate buffer containing 0.2% formic acid (30:70, v/v) with a flow rate of 0.3 mL/min.
3.2. Conditions for ESI-MS
Due to amino and carboxylic groups in its chemical structure,cefcapene acid had mass spectrometric response either in the positive ion mode or in the negative ion mode. The test result showed that the signal intensity of the analyte obtained in the positive mode was much higher than that in the negative mode. So, the positive monitoring mode was selected in the MS detection. The protonated molecular ion [M+H]+at m/z 454.2 was selected as the target ion for cefcapene acid.Comparing with capillary voltage, desolvation gas (drying gas)flow and temperature,fragmentor voltage is considered to be the most important instrumental parameter in the optimization of ionization settings [15,16]. The intensity of this ion was compared at fragmentor voltages of 70, 90, 100, 120, 140 and 160 V. The result showed that the highest sensitivity was obtained using a fragmentor voltage of 120 V. More detailed optimizing was conducted at fragmentor voltages of 115, 120,125, 130 and 135 V. The result showed that the highest sensitivity was obtained using a fragmentor voltage of 125 V.Therefore, a fragmentor voltage of 125 V was used to carry out LC-ESI-MS in the SIM mode.At this fragmentor voltage the most intensive ion of the IS was the ion [M+H]+at m/z 348.2. Therefore, the positive ion [M+H]+at m/z 348.2 was selected as the target ion for the IS (see Fig.2).

Fig.2 Mass spectra of the positive ions of cefcapene acid(A) and cefalexin (B) at 125 V fragmentor voltage.
3.3. Sample preparation
In the method for determination of cefcapene acid in human plasma, the plasma sample was precipitated with methanol and then the extracts were injected into the mobile phase stream without evaporation and reconstitution. It could simplify the sample preparation procedure significantly and also meet the requirements of the assay. No interference was observed from any endogenous or exogenous plasma matrix.
3.4. Selectivity
Selectivity was assessed by comparing the chromatograms of drug-free human plasma and urine with the corresponding spiked plasma and urine, for the test of endogenous interferences. For plasma, observed retention time for cefcapene acid and the IS was 2.5 and 3.2 min, respectively. For urine,observed retention time for cefcapene acid and the IS was 2.9 and 3.9 min, respectively. The typical chromatograms of the plasma and urine blank samples are shown in Figs. 3(A) and 4(A). No peak of the endogenous substances was observed,which would interfere with the detection of cefcapene acid and the IS.Figs.3(B)and 4(B)show the chromatograms of spiked plasma and urine samples with cefcapene acid at LLOQ level and the IS. Typical chromatograms of plasma and urine samples are shown in Figs. 3(C) and 4(C), which were collected 0.75 h after oral administration of a single dose of 50 mg cefcapene pivoxil hydrochloride tablet for plasma and 2-4 h after oral administration of a single dose of 100 mg cefcapene pivoxil hydrochloride tablet for urine, respectively.The corresponding concentrations of cefcapene acid in plasma and urine were found to be 0.6458 and 108.0 μg/mL,respectively.

Fig.3 Typical SIM chromatograms obtained from (A) blank plasma sample, (B) plasma spiked with cefcapene acid at the LLOQ level (0.03 μg/mL) and with the IS, and (C) plasma obtained from a Chinese volunteer 0.75 h after oral administration of 50 mg cefcapene pivoxil hydrochloride tablet.

Fig.4 Typical SIM chromatograms obtained from (A) blank urine sample, (B) urine spiked with cefcapene acid at the LLOQ level (0.1 μg/mL) and with the IS, and (C) urine obtained from a Chinese volunteer 2-4 h after oral administration of 100 mg cefcapene pivoxil hydrochloride tablet.
3.5. Linearity and LLOQ
The validated assays were linear in the range from 0.03 to 5 μg/mL for plasma and from 0.1 to 400 μg/mL for urine,respectively.The typical calibration curves of cefcapene acid in human plasma and urine were Y=0.7390C+0.009650 and Y=0.01315C+0.0004304, respectively, with both coefficients of correlation (r) about 0.999. The lowest concentration of determination with RSD<20%was taken as the LLOQ which was found to be 0.03 μg/mL for plasma and 0.1 μg/mL for urine. In all the cases, the calculated concentrations in the calibration curves were within ±15% bias from the nominal value except at the LLOQ, which was set at ±20%.
3.6. Precision and accuracy
Table 1 summarizes the intra- and inter-day precision and accuracy for cefcapene acid evaluated by assaying the QC samples.The precision was calculated using one-way ANOVA.The results demonstrated that the values were within the acceptable range and the methods were accurate and precise [17].
3.7. Recovery and matrix effect
The observed value of extraction recovery of the methods from plasma and urine and %RSD (n=5) are shown in Table 2. Recoveries were more than 80% at different QC concentrations for both plasma and urine with acceptable variability.
Matrix interferences caused by plasma and urine endogenous materials were evaluated by comparing the peak areas of the post-spiked standards with those of the neat standards at the QC concentrations, the ratio was within the acceptable limits (85-115%). No significant ion suppression or enhancement was observed at the expected retention time of the targeted ions.

Table 1 Precision and accuracy of the LC-MS method to determine cefcapene acid in human plasma and urine (in three analysis batch, five replicates for each batch).

Table 2 Results of extraction recovery of cefcapene acid in human plasma and urine (n=5).

Table 3 Summary of stability of cefcapene acid in human plasma under different storage conditions(n=3).
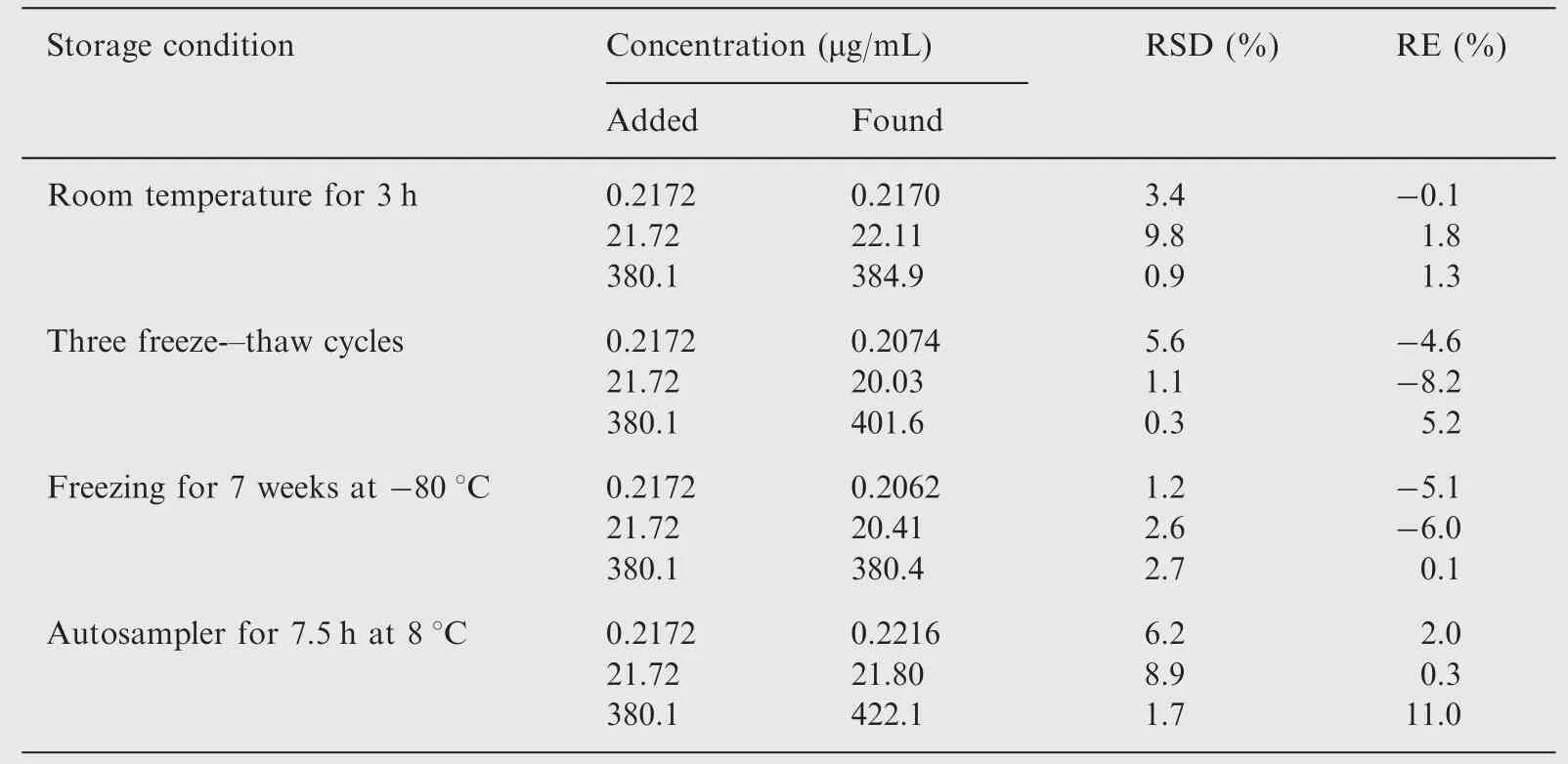
Table 4 Summary of stability of cefcapene acid in human urine under different storage conditions(n=3).

Fig.5 The mean plasma concentration-time curves of cefcapene acid in healthy Chinese volunteers (n=12) after single oral administration of 50, 100 and 200 mg cefcapene pivoxil hydrochloride tablet, respectively.

Fig.6 The mean urinary recovery-time curve of cefcapene acid in healthy Chinese volunteers (n=12) after single oral administrations of 100 mg cefcapene pivoxil hydrochloride tablet.
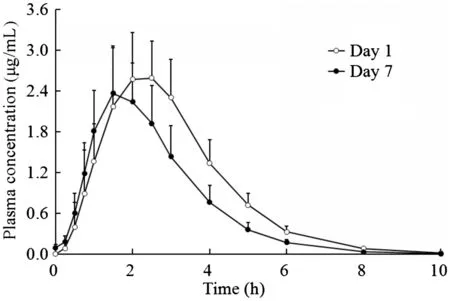
Fig.7 The mean (±SD) plasma concentration-time curves of cefcapene acid in healthy Chinese volunteers (n=12) after multiple oral administrations of 100 mg cefcapene pivoxil hydrochloride tablets.
3.8. Stability
The results obtained from determination of stability (Tables 3 and 4) showed no significant degradation occurred under the conditions tested. The plasma samples were stable at ambient temperature for 3.5 h, during three freeze-thaw cycles, and at-80°C for 10 weeks. The urine samples were stable at ambient temperature for 3 h, during three freeze-thaw cycles, and at-80°C for 7 weeks. The stock solutions of cefcapene acid and the IS in methanol were observed to remain stable for 1 month at-20°C.
3.9. Application of the method in pharmacokinetic studies
3.9.1. Single-dose pharmacokinetics
All the 12 enrolled subjects participated in three phases of the PK study. The mean plasma concentration-time curves of cefcapene acid after a single oral dose of 50, 100 and 200 mg are shown in Fig.5, and the pharmacokinetic parameters are summarized in Table 5.Over the dose range studied,the mean Cmax,AUC0-tand AUC0-∞increased linearly across the doses by linear regression analysis. Dose proportionality was observed (P>0.05) by the analysis of ANOVA on the values of ln(Cmax/dose) and ln(AUC/dose) among the three dose groups. Thus, Cmaxand AUC proved to be dose proportional across the studied doses by different methods. The value of Tmaxincreased slightly with dose escalation.The values of t1/2,CL/F and Vd/F were independent of dose(P>0.05).The Cmaxand AUC were higher than literature reported [5] which indicated that cefcapene pivoxil hydrochloride tablets may exist racial difference.
The urinary recovery of cefcapene acid versus time after oral administration of the drug to the human subjects is shown in Fig.6.The cumulated rate of cefcapene acid excreted in urine was 60.1%,which is also higher than literature reported [6].
3.9.2. Multiple-dose pharmacokinetics
The 12 subjects received 100 mg cefcapene pivoxil hydrochloride tablets thrice daily for 7 days. The mean plasma concentrationtime curves of cefcapene acid after the first dose (day 1) and the last dose(day 7)are presented in Fig.7,and the pharmacokinetic parameters from the non-compartmental analysis of measured plasma concentrations on day 1 and day 7 are provided in Table 5. Mean (SD) plasma concentrations (Cmin-ss) on the 4th,5th, 6th, and 7th days before dosing were 0.08108 (0.03988),0.1022(0.03782),0.07065(0.03098)and 0.09925(0.04501)μg/mL,respectively. No significant difference in Cmin-sswas found by ANOVA analysis, indicating that steady state was achieved after administration of 100 mg cefcapene pivoxil hydrochloride tablets for 7 consecutive days. Under steady state conditions, cefcapene acid was absorbed with the median Tmaxof 1.7 h and a mean Cmaxof 2.466 μg/mL.And cefcapene acid cleared from plasma with no significant difference of t1/2between the first and the last dose.But,the mean AUC values were lower in multiple-dosing regimen than the corresponding values obtained after single-dose (day 1)administration, and no accumulation was found following repeat dosing of cefcapene acid with Rac of 0.76 for AUC0-10.A high DF of cefcapene acid in plasma was achieved at 2.8.
4. Conclusion
The simple, rapid and specific LC-ESI-MS methods for quantification of cefcapene acid in human plasma and urine have been developed and validated. There were no significant interferences from endogenous compounds. The fully validated methods have been successfully applied to the pharmacokinetics in health Chinese volunteers.
[1] K. Ishikura, T. Kubota, K. Minami, et al., Synthesis and structure-activity relationships of 7 beta-[(Z)-2-(2-aminothiazol-4-yl)-3-(substituted)-2-propenoyl-amino]-3- cephems with C-3 substitutions, J. Antibiot. 47 (1994) 466-476.
[2] A. Saito, New antimicrobial agent series L: cefcapene pivoxil,Jpn. J. Antibiot. 6 (1997) 485-506.
[3] H. Bundgaard, U. Klixbull, Hydrolysis of pivampicillin in buffer and plasma solutions. Formation of a 4-imidazolidinone from ampicillin and formaldehyde, Int. J. Pharm. 27 (1985) 175-183.
[4] M. Johansen, H. Bundgaard, E. Falch, Spectrophotometric determination of the rates of hydrolysis of aldehyde releasing prodrugs in aqueous solution and plasma, Int. J. Pharm. 13(1982) 89-98.
[5] I. Saikawa, Y. Nakajima, M. Tai, et al., Studies on beta-lactam antibiotics for medicinal purpose.XXII.Studies on the metabolism of pivaloyloxymethyl(6R,7R)-7-[(Z)-2-(2-amino-thiazol-4-yl)-2-methoxy-iminoacetamido]-3-[(5-methyl-2H-tetrazol-2-yl)methyl]-3-cephem-4-carb-oxylate(T-2588) (2)], Yakugaku Zasshi 106 (1986) 478-490.
[6] K.Totsuka,K.Shimizu,M.Konishi,et al.,Metabolism of S-1108,a new oral cephem antibiotic, and metabolic profiles of its metabolites in humans, Antimicrob. Agents Chemother. 36 (1992) 757-761.
[7] Y.Toyonaga,T.Ishihara,T.Tezuka,et al.,Pharmacokinetic and clinical studies of S-1108 in the pediatric field,Jpn.J.Antibiot.46(1993) 991-1002.
[8] R. Fujii, T. Abe, T. Tajima, et al., Pharmacokinetic and clinical studies of S-1108 in the pediatric field. Pediatric Study Group of S-1108, Jpn. J. Antibiot 48 (1995) 921-941.
[9] M. Fujimoto, Pharmacokinetics of cefcapene pivoxil and AS-924 in gastrectomized patients, Int. J. Antimicrob. Agents 18 (2001)489-494.
[10] K. Mizojiri, S. Futaguchi, R. Norikura, et al., Disposition of S-1108, a new oral cephem antibiotic, and metabolic fate of pivalic acid liberated from [pivaloyl-14C]S-1108 in rats and dogs,Antimicrob. Agents Chemother. 39 (1995) 1445-1453.
[11] M. Nakashima, T. Uematsu, T. Oguma, et al., Phase I clinical studies of S-1108: safety and pharmacokinetics in a multipleadministration study with special emphasis on the influence on carnitine body stores, Antimicrob. Agents Chemother. 36 (1992)762-768.
[12] US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research.Guidance for Industry, Bioanalytical Method Validation, May 2001.
[13] State Food and Drug Administration, The Guidance of Clinical Pharmacokinetics Study Technique for Chemistry Drug in Human (No. [H] GCL1-2).
[14] S.A. Parekh, A. Pudage, S.S. Joshi, et al., Rapid and sensitive liquid chromatography-tandem mass spectrometry(LC-MS/MS)method for the determination of clonidine in human plasma,J. Chromatogr. B 867 (2008) 172-178.
[15] W.J. Zhou, L. Ding, Y.Q. Wang, et al., Solid phase extraction and liquid chromatography-electrospray ionization-mass spectrometry for the determination of bencycloquidium bromide in human plasma, J. Chromatogr. B 877 (2009) 897-901.
[16] W.J. Zhou, L. Ding, G. Xu, et al., Determination of bencycloquidium bromide in human urine using weak cation-exchange solid-phase extraction and LC-ESI-MS: method validation and application to kinetic study of urinary excretion, J. Pharm.Biomed. Anal. 50 (2009) 35-40.
[17] C.C. Chan, H. Lam, Y.C. Lee, et al., Analytical Method Validation and Instrument Performance Verification, John Wiley& Sons Inc., Hoboken, 2004.
 Journal of Pharmaceutical Analysis2013年2期
Journal of Pharmaceutical Analysis2013年2期
- Journal of Pharmaceutical Analysis的其它文章
- Analysis of spironolactone residues in industrial wastewater and in drug formulations by cathodic stripping voltammetry
- Anodic voltammetric determination of gemifloxacin using screen-printed carbon electrode
- Electrochemical study and application on rutin at chitosan/graphene films modified glassy carbon electrode
- Liquid chromatography tandem mass spectrometry method for the estimation of lamotrigine in human plasma:Application to a pharmacokinetic study
- New simple spectrophotometric method for determination of the binary mixtures (atorvastatin calcium and ezetimibe;candesartan cilexetil and hydrochlorothiazide) in tablets
- Establishment of inherent stability of pramipexole and development of validated stability indicating LC-UV and LC-MS method