
儿童原发性1 型高草酸尿症1 例并文献复习
2013-12-23 04:23李国民刘海梅
中国循证儿科杂志 2013年6期
李国民 沈 茜 徐 虹 孙 利 安 宇 刘海梅 曹 琦
原发性1 型高草酸尿症(Primary hyperoxaluria type 1,PH 1;OMIM # 259900)为罕见的乙醛酸盐代谢异常性疾病,为常染色体隐性遗传[1,2]。因肝脏特异的丙氨酸乙醛酸转氨酶(alanine/glyoxylate aminotransferase,AGT,EC 2.6.1.44)缺陷所引起,AGT 由AGXT 基因编码。由于肝内AGT缺乏,乙醛酸转氨生成甘氨酸减少,而氧化生成草酸增加,导致草酸盐(主要为草酸钙)在多个器官组织沉积[3,4]。PH 1 主要的临床特征为多发性和复发性肾脏结石和(或)肾钙质沉着症[5]。在欧洲,PH 1 患病率为1 ~3/100 万,活产婴儿中的发病率为1/12 万[3,6,7]。约有50%PH 在儿童期起病,15 岁前进展为终末期肾病(ESRD)[8,9]。由于对PH 1 认识不足常误诊和漏诊,未能及时或不恰当治疗使患儿过早进入ESRD[1,10,11]。截止目前,中国台湾和香港地区各有1 例儿童期起病的PH 1 报道,大陆地区有4 例儿童期起病的PH 1 报道,但均未对AGXT 基因进行分析[12~16]。本文报道1 例确诊的PH 1 患儿临床特征和AGXT 基因分析结果。
1 病例报告
患儿,女,汉族,2002 年8 月出生,2012 年2 月复旦大学附属儿科医院(我院)门诊以“慢性肾脏病(3 期)、双肾及右输尿管结石和右肾积水”收住院。
患儿于2005 年8 月无明显诱因下出现肉眼血尿,外院肾脏B 超提示肾脏多发性小结石,无肾盂扩张,当时未予特殊处理。2007 年3 月患儿出现右侧腰、背部疼痛,外院肾脏B 超提示双肾多发结石伴右肾积水,予经皮肾镜取石、右肾体外震波碎石及短期口服排石药物治疗,半年后复查B 超,仍然提示双肾结石,但无积水,后未随访。2012 年1 月再次因右侧腰、背部疼痛就诊当地医院,肾脏B 超提示双肾多发性结石,右侧输尿管上端结石,伴右肾积水,当地医院再行震波碎石术治疗,术后右侧输尿管放置双J 管(DJ 管),住院期间SCr 185 μmol·L-1,腹部X 线平片提示双肾多发性结石,右侧输尿管上端结石,伴右肾积水,99mTc-DTPA(二乙三胺5 醋酸)肾动态显像检查提示左肾功能轻中度受损,右肾功能中重度受损,双肾肾小球滤过率(GFR,mL·min-1·1.73m-2)均低于正常(左肾22,右肾7.7,双肾39.7)。
入我院查体:神智清楚,体重33 kg(同年龄、同性别正常人群的P50),身高136 cm(同年龄、同性别正常人群的P25~P50);血压98/60 mmHg。除右肾区叩击痛外,未发现其他阳性体征。家族史:患儿父母非近亲结婚,其母系及父系3 代28 人均无相同疾病史。病程中患儿无头痛、呕吐,尿量减少不明显,精神、食欲尚可,生长发育未见明显异常。
实验室检查:血常规:Hb 119.0 g·L-1,RBC 4.25 ×1012·L-1,WBC 7.2 ×109·L-1,PLT 197 ×109·L-1。尿常规:RBC 2 ~3·HP-1,WBC 25 ~30·HP-1,潜血(+ ++),蛋白微量。尿生化未见异常。血生化:ALT 9 U·L-1,AOT 15 U·L-1,白蛋白44.9 g·L-1,球蛋白23.5 g·L-1,总胆固醇4. 28 mmol·L-1,三酰甘油1. 60 mmol·L-1,BUN 13.20 mmol·L-1,SCr 155.0 μmol·L-1,尿酸389.0 μmol·L-1,甲状旁腺素107.0 pg·L-1。
影像学检查:肾脏B 超提示双肾内多发性结石,右肾盂增宽,双侧输尿管无扩张,腹部X 线平片(图1A)和CT(图1B ~D)均提示双肾多发性结石。肝脏、脾脏B 超正常,心脏超声正常,双手X 线平片正常。
结石成份分析为单水草酸钙。
基因分析:对患儿及其父母进行AGXT 基因外显子及其附近区域进行直接测序,AGXT 基因分析发现,患儿为杂合突变c. 242C >A(p. Ser81X)和c. 823_824dupAG(p.Ser275delinsArgAlafs),其父亲携带c.242C >A,其母亲携带c.823_824dupAG(图2)。100 例正常健康人未发现c.242C>A 和c.823_824dupAG。
经我院肾脏和泌尿系统疾病诊治中心(泌尿外科、肾内科及影像科)讨论诊断为①PH 1,②慢性肾脏病(3 期)。嘱患儿大量饮水(每日3 L 以上)、碱化尿液和口服维生素B6治疗。
2012 年9 月20 日再次入我院,由于患儿不能坚持上述治疗,肾功能持续恶化,BUN 16.20 mmol·L-1,SCr 680.5 μmol·L-1,予血液透析治疗。目前患儿在我院血液透析,每周3 次,透析充分性好(Kt/V 1. 49,尿素清除率70%),血压平稳,Hb 维持在120 g·L-1左右,血钙和血磷正常,甲状旁腺素维持在300 pg·L-1左右;定期行心脏超声、四肢X 线平片等检查,现患儿未发现草酸钙在以上器官沉积,等待器官移植。
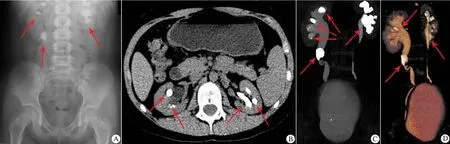
图1 双肾影像学检查所见Fig 1 Renal imaging findings
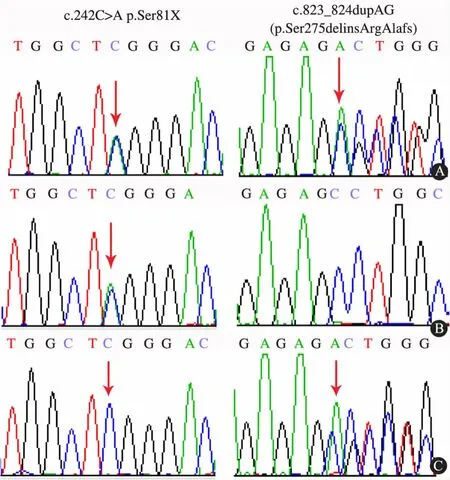
图2 家系及AGXT 基因分析图Fig 2 Family tree andAGXT gene analysis
2 讨论
PH 从分子水平可将其分为PH 1、PH 2 和PH 3 三种类型[1]。PH 1 是由AGXT 基因突变引起,突变导致维生素B6依赖的肝脏特异的AGT 缺失或功能异常。PH 2 是由GRHPR 基因突变引起,突变导致乙醛酸/羟基丙酮酸还原酶功能缺陷。PH 3 是由HOGA1 基因突变引起,突变导致线粒体4-羟基-2-酮戊二酸醛缩酶功能异常[1,4]。3 种类型的PH 均导致肾脏草酸钙结石病,其中PH 1 最为常见,约占PH 的80%[1,11]。
在欧洲、北美地区,PH 1 的年发病率为0.1 ~0.2/100万,患病率为0.8 ~2.9/100 万。欧洲活产婴儿中PH 1 的发病率为1/10 ~12 万[7,10]。在欧洲、美国和日本,约1%的儿童ESRD 是由PH 1 引起[1,2,10]。近亲结婚较多的中东和北非地区PH 1 的发病率更高,如突尼斯报道PH 1 的发病率达到5.5/100 万,13%的儿童ESRD 是由PH 1 引起,而科威特报道也有约10%的儿童ESRD 是由PH 1 引起[11,17,18]。国内目前尚无基于人群的PH 1 发病率的报道[12 ~14]。
任何年龄的人群均可发生PH 1。PH 1 的临床表现异质性很大,可无任何临床症状,有的表现血尿、腹痛,有的甚至就诊时表现为ESRD[1,19]。同一家族患者间的临床表型差异也很大,是造成PH 1 漏诊和误诊的主要原因之一[2,19]。PH 1 主要特征为复发性肾结石和肾钙质沉着症。早期可能只有血浆草酸浓度增加和尿草酸盐排出增加,随着结石形成可出现血尿和腹痛。结石还可引起泌尿道感染和梗阻性肾病等并发症。肾钙质沉着症和结石形成均能引起肾功能损害,出现进行性的肾功能恶化,最后导致慢性肾功能衰竭。当肾功能受损,GFR <40 mL·min-1·1.73 m-2,排泄草酸的能力进一步下降,体内草酸蓄积更为明显,进而沉积于肾脏以外的组织器官,从而发生相应损害,即出现草酸盐沉积症,可累及骨骼(导致骨痛、骨钙化、骨关节畸形、病理性骨折,累及骨髓时导致贫血,促红细胞生成素治疗无效),心脏(心肌病、传导阻滞、低血压),血管(播散性、闭塞性血管病灶,肢体坏疽,内瘘栓塞),神经系统(周围神经病变、单神经炎、多神经炎),皮肤(溃疡、网状青斑),视网膜病变,肝脾增大,睾丸增大和淋巴结增大等[1,10,11,20]。
辅助检查有助于PH 1 的诊断。①尿草酸水平测定:在疾病早期无任何临床症状时,尿草酸排出增加,典型的PH 1患者的尿草酸浓度>1 mmol·1.73 m-2(正常<0.5 mmol·1.73m-2)[4]。②血浆草酸水平测定:疾病早期血浆草酸浓度并不升高,当GFR <60 mL·min-1·1.73 m-2时,血浆草酸浓度才升高,GFR <40 mL·min-1·1.73 m-2时,血浆草酸浓度可迅速升高>30 μmol·L-1(正常为1 ~6 μmol·L-1)[17],血、尿草酸测定受人为因素影响较大,目前临床几乎不开展这两个项目;尿液草酸水平上升对PH 1早期诊断有提示作用,但需要排除胃肠疾病所引起的血、尿草酸水平上升,如炎症性肠病、短肠综合征等[2,10,11]。③结石成份分析:PH 1 的结石成份95%为单水草酸钙,为特征性改变[11];结石成份分析有重要的诊断价值,而且操作简单。④影像学检查:腹部CT、X 线平片和B 超均可发现双肾多发性结石,CT 还能发现肾髓质钙质沉着症[1,10,11],而X 线平片和B 超不能发现肾钙质沉着症[11,21]。⑤AGXT基因分析:AGXT 外显子及其剪切区直接测序是诊断PH 1的重要和有效的手段[11,22],如AGXT 基因分析发现致病性纯合或复合杂合突变就可以确诊[24,25]。⑥肝脏穿刺活检:PH 1 诊断的金标准[22,23],可以分析AGT 活性、AGXT 基因的mRNA 和蛋白表达水平。由于肝穿刺是一个创伤性检查,其作用正逐渐被AGXT 基因分析所替代,目前只有在AGXT 基因分析不能解释疾病时才考虑肝穿刺检查。
1990 年,AGXT 基因cDNA 序列被首次克隆。AGXT 基因含有11 个外显子,位于常染色体2q37.3(GenBank NT_005416),其编码AGT。AGT 由392 个氨基酸构成,主要在肝细胞过氧化酶体中产生。单体型研究发现人类存在主要等位基因(major alleles,AGXT-Ma)和次要等位基因(minor alleles,AGXT-Mi),在白种人中分布频率分别为80% 和20%[24,26,27]。在日本和中国人群中,AGXT-Mi 频率只有2%。与AGXT-Ma 不同的是,AGXT-Mi 在编码区有p. P11L和p. I340M 两个变异,以及内含子1 上存在一个含有74个碱基重复变异,这3 个变异均为多态性[24,25]。尽管这3个变异并不致病,但它们使得AGT 更易受AGXT 错义突变影响。主要和次要等位基因均可发生突变,引起疾病。到目前为止,超过150 个致病性AGXT 突变被报道,这些突变包括无义突变、错义突变、框移突变和剪切突变等[1,5,28]。AGXT 基因的11 外显子均可发生突变,c. 508G >A(p.G170R)、c.33_34insC、c.731T >C 和c.454T >A 是最常见的4 个PH 1 致病突变位点,其中c.508G >A 位于AGXT-Mi上[29]。既往将外显子1、4 和7 作为靶向序列进行分析,只能发现70%肝脏病理证实的PH 1 存在AGXT 基因突变,故整个AGXT 基因编码区及其剪切区序列分析能增加基因诊断的敏感性[1,2,17]。
本例患儿临床表现符合PH 的临床特点,结石成份分析为单水草酸钙,基因分析提示存在AGXT 基因的c.242C>A 和c.823_824dupAG 突变。而c.823_824dupAG 是已报道的致病突变,c.242C >A 为无义突变,可导致AGT 蛋白被部分编码,对AGT 的功能有严重的影响,并且100 例正常健康人群未发现有c.242C >A 突变,故c.242C >A 也是致病性突变。据此可认为患儿为AGXT 基因复合杂合突变。结合临床表现和相关检查,PH 1 诊断明确,无需肝穿刺检查。
PH 1 的治疗主要为保守治疗,包括大量饮水、口服维生素B6、碱化尿液和饮食治疗。大量饮水是指每天饮水量超过3 L·m-2,饮水次数均匀分布在24 h 内。婴幼儿不能耐受可考虑鼻饲或胃造篓术。如果出现腹泻、呕吐和发热,以及外科限制口服液体等情况时,应该考虑静脉补液[11,17]。维生素B6是AGT 的辅酶,口服维生素B6可使10% ~30% 的PH 1 患者的尿草酸盐下降。开始剂量5 mg-1·kg-1·d-1,逐步增加至最大剂量20 mg-1·kg-1·d-1[11]。碱性尿液能增加草酸钙的溶解饱和度,减轻结石增大和肾髓质草酸钙质沉着,故对PH 1 患者可给予柠檬酸钾口服0.1 ~0.3 mg-1·kg-1·d-1[10,11]。限制富含草酸的食物,如禁食菠菜、木耳、浓茶和巧克力等。但不限制钙的食入,饮食钙的限制会增加肠道草酸的吸收。当出现多发性结石,以及因其引起的感染和尿道梗阻时需要外科的干预。可考虑经皮肾镜取石,不主张体外震波碎石和开放性手术取石。如患者有反复肾绞痛,可考虑放置双J 管。对已行肾脏替代治疗的患者,有时可考虑双侧肾切除以减少感染和梗阻等问题[5]。随着PH 1 病情进展,应该首先考虑器官移植,器官移植时应该采用肝肾联合移植,不主张单独肾脏移植[19,30,31]。肝肾联合移植应该在进展至慢性肾脏病4 期前进行,避免草酸盐在其他器官沉积[11,32]。这与其他原因引起的慢性肾脏病5 期进行肾脏替代治疗不同。当不便行肝肾联合移植时,又有透析指证时可选择高效的透析治疗,血液透析、腹膜透析或两者的联合均可,而血液透析对草酸的清除要好于腹膜透析[11,17]。本例患儿早期的诊治医生对PH 1 认识不足和患儿对治疗耐受性差是其肾功能快速恶化的主要原因。
[1]Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med,2013,369(7):649-658
[2]Hoppe B, Beck BB, Milliner D S. The primary hyperoxalurias. Kidney Int,2009,75(12):1264-1271
[3]Cochat P, Fargue S, Bacchetta J, et al. Primary hyperoxaluria. Nephrol Ther,2011,7(4):249-259
[4]Chand AQ, Kaskel FJ. Pediatrics: Timely diagnosis of primary hyperoxaluria type 1. Nat Rev Nephrol,2009,5(12):670-671
[5]Edvardsson VO, Goldfarb DS, Lieske J C, et al. Hereditary causes of kidney stones and chronic kidney disease. Pediatr Nephrol,2013,28(10):1923-1942
[6]Jungers P, Joly D, Blanchard A, et al. Inherited monogenic kidney stone diseases: recent diagnostic and therapeutic advances. Nephrol Ther,2008,4(4):231-255
[7]Belhaj R, Hayder N, Gargueh T, et al. Biochemical and molecular diagnosis of primary hyperoxaluria type 1: Tunisian study about 15 cases. Pathol Biol (Paris),2011,59(4):e97-e102
[8]Saner FH, Treckmann J, Pratschke J, et al. Early renal failure after domino liver transplantation using organs from donors with primary hyperoxaluria type 1. Transplantation,2010,90(7):782-785
[9]Jungers P, Joly D, Barbey F, et al. ESRD caused by nephrolithiasis: prevalence, mechanisms, and prevention. Am J Kidney Dis,2004,44(5):799-805
[10]van der Hoeven SM, van Woerden CS, Groothoff JW. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults:results of the Dutch cohort. Nephrol Dial Transplant,2012,27(10):3855-3862
[11]Cochat P, Groothoff J. Primary hyperoxaluria type 1: practical and ethical issues. Pediatr Nephrol,2013,28(12):2273-2281
[12]Zhou JH(周建华),Cui W,Wang YQ. 儿童原发性高草酸尿症致双肾广泛结石、钙化和肾功能衰竭一例. Chin J Nephrol(中华肾脏病杂志),2004,20(1):46
[13]Chen Z(程震),Tang Z,Chen HP, et al. Literature review of two misdiagnosed patients with primary hyperoxaluria. Clinical Misdiagnosis & Mistherapy(临床误诊误治),2013,26(2):18-21
[14]Wu HW(伍汉文), Wang YL. 原发性高草酸尿症一例.Chin J Pediatr(中华儿科杂志),1994,32(2):88
[15]Wong PN, Tong MW, Mak SK, et al. Late-onset primary hyperoxaluria type 1 in a Chinese individual with absent alanine: glyoxylate aminotransferase activity. Chin Med J(Engl),2004,117(12):1889-1890.
[16]Wong PN, Law EL, Tong GM, et al. diagnosis of primary hyperoxaluria type 1 by determination of peritoneal dialysate glycolic acid using standard organic-acids analysis method.Perit Dial Int,2003,23(S2):210-213
[17]Cochat P, Hulton SA, Acquaviva C, et al. Primary hyperoxaluria Type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant,2012,27(5):1729-1736
[18]Sanjad SA, Al-Abbad A, Al-Sabban E. Primary hyperoxaluria type 1: An underestimated cause of nephrocalcinosis and chronic renal failure in Saudi Arabian children. Ann Saudi Med,1999,19(1):4-7
[19]Heffron TG, Rodriguez J, Fasola CG, et al. Successful outcome after early combined liver and en bloc-kidney transplant in an infant with primary hyperoxaluria type 1: a case report. Pediatr Transplant,2009,13(7):940-942
[20]Beck BB, Habbig S, Dittrich K, et al. Liver cell transplantation in severe infantile oxalosis--a potential bridging procedure to orthotopic liver transplantation?. Nephrol Dial Transplant,2012,27(7):2984-2989
[21]Raju DL, Cantarovich M, Brisson ML, et al. Primary hyperoxaluria: clinical course, diagnosis, and treatment after kidney failure. Am J Kidney Dis,2008,51(1):e1-e5
[22]Williams EL, Acquaviva C, Amoroso A, et al. Primary hyperoxaluria type 1: update and additional mutation analysis of the AGXT gene. Hum Mutat,2009,30(6):910-917
[23]Hoppe B. Evidence of true genotype-phenotype correlation in primary hyperoxaluria type 1. Kidney Int,2010,77(5):383-385
[24]Nagara M, Tiar A, Ben HN, et al. Mutation spectrum of primary hyperoxaluria type 1 in Tunisia: implication for diagnosis in North Africa. Gene,2013,527(1):316-320
[25]Robbiano A, Mandrile G, De Marchi M, et al. Novel human pathological mutations. Gene symbol: AGXT. Disease:hyperoxaluria. Hum Genet,2010,127(4):468
[26]Cellini B, Montioli R, Paiardini A, et al. Molecular Insight into the Synergism between the Minor Allele of Human Liver Peroxisomal Alanine:Glyoxylate Aminotransferase and the F152I Mutation. J Biol Chem,2009,284(13):8349-8358
[27]Coulter-Mackie MB, Applegarth D, Toone JR, et al. The major allele of the alanine:glyoxylate aminotransferase gene:seven novel mutations causing primary hyperoxaluria type 1.Mol Genet Metab,2004,82(1):64-68
[28]Kanoun H, Jarraya F, Hadj SI, et al. A double mutation in AGXT gene in families with primary hyperoxaluria type 1.Gene,2013,531(2):451-456
[29]Fargue S, Lewin J, Rumsby G, et al. Four of the most common mutations in primary hyperoxaluria type 1 unmask the cryptic mitochondrial targeting sequence of alanine:glyoxylate aminotransferase encoded by the polymorphic minor allele. J Biol Chem,2013,288(4):2475-2484
[30]Mor E, Nesher E, Ben-Ari Z, et al. Sequential liver and kidney transplantation from a single living donor in two young adults with primary hyperoxaluria type 1. Liver Transpl,2013,19(6):646-648
[31]Scheinman JI. Liver transplantation in oxalosis prior to advanced chronic kidney disease. Pediatr Nephrol,2010,25(11):2217-2222
[32]Harps E, Brinkert F, Ganschow R, et al. Immediate postoperative intensive care treatment of pediatric combined liver-kidney transplantation: outcome and prognostic factors.Transplantation,2011,91(10):1127-1131
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
西北大学学报(自然科学版)(2021年3期)2021-06-28
肾脏病与透析肾移植杂志(2021年1期)2021-01-13
健康体检与管理(2021年10期)2021-01-03
中国保健营养(2019年7期)2019-10-21
中成药(2017年7期)2017-11-22
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
健康管理(2016年8期)2016-05-14
现代养生·上半月(2015年11期)2015-12-08
