
新型藏药愈心安神滴丸的制备工艺研究
2013-11-29 08:01马文俊李宝文东知多杰
中国民族医药杂志 2013年7期
马文俊 李宝文 贾 明 东知多杰
(青海阿如拉藏医药研究开发有限公司,青海 西宁 810003)
本项目主要目标是研制一种治疗和预防由气滞血瘀所引起的疾病的藏药新产品愈心安神滴丸,其核心研究内容主要是药物的功效、安全性以及制剂的剂型。制备关键工艺技术主要是原药材的加工炮制技术、挥发油提取工艺技术、挥发油包合工艺技术及滴丸的成型工艺技术;确定生产工艺的稳定性、可操作性及质量控制要求。
1 材料、设备
1.1 处方组成:广枣3000g,当归150g,干姜150g,肉豆蔻150g,诃子 150g,寒水石 150g。
1.2 主要实验设备和检验仪器:磁力搅拌器、高速粉碎机、滴丸机。
1.3 制法概述:以上六味,除寒水石外,其余药材提取挥发油,用β-环糊精包合,备用;药渣加水煎煮三次,滤过,合并滤液,滤液浓缩成相对密度为1.26~1.28(50℃)的清膏,加乙醇使含醇量达65%,搅匀,静置24h,取上清液,回收乙醇,浓缩干燥,粉碎并过100目筛,备用;寒水石研细并过200目筛。取聚乙二醇4000与聚乙二醇6000适量,加热使溶融,加入上述挥发油包合物、稠膏和寒水石细粉,混匀,滴入冷却的液体石蜡中,制成1000g滴丸,即得。
1.4 工艺路线设计[1-2]
1.4.1 处方中主要有广枣、当归、干姜、肉豆蔻、诃子和寒水石六味药材,鉴于其中许多药材含有挥发油,因此需要先提取挥发油,采用直接水浸泡后煮沸提取,冷凝收取挥发油。
1.4.2 方中寒水石为矿物类药材,配方时需要进行奶制,主要成分为碳酸及硫酸钙,在水中基本不溶,不宜采用提取方法。因此选择粉碎处理后直接加入滴丸成型过程的方法;其余均为植物性药材的果实类和根类,适合用提取的方法。
1.4.3 因原方为口服散剂,疗效确定,所以提取方法采用水煮提,水提取后的提取物中有比较多的杂质(多糖、鞣质等),因此提取浓缩后应进行乙醇精制处理,以改善外观和减少基质用量,达到外观适当和减少用量的目的。
1.4.4 挥发油处理:由于滴丸制备时需要在比较高的温度下进行,直接加入时挥发性成分容易损失,所以应采用保护技术,设计采用β-环糊精包合方式,在成型时以固体粉末形式和不提取的寒水石一起直接加入滴丸成型环节。
1.4.5 根据资料检索得知,聚乙二醇是水溶性人体不吸收的常用基质,是目前在制剂中使用比较多的滴丸基质,初步选择聚乙二醇(PEG)为首选基质。资料表明采用PEG4000和PEG6000加工的滴丸比较理想,本试验同样选择上述基质进行研究,具体配合比例及用量通过实验确定。
1.5 愈心安神滴丸工艺流程图:

2 关键工艺技术实验研究方法
2.1 剂型选择:愈心安神滴丸是在尚未完全了解组方有效成分前提下开展的科研项目,由于复方制剂有效成分的研究仍然存在困难,提取过程中代表性成分的选择不易确定,在保留传统藏药特点的基础上结合现代药品剂型的发展要求,选用滴丸为最终制剂剂型。
2.2 挥发油提取工艺的筛选和确定:基于研究中药药性、提高药物疗效、改进制剂等目的,需要将有效成分从药材中提取分离出来。
为提高提取效率,需要对影响“传质”的因素进行认真考虑,这些因素包括药材的浸泡时间、提取的溶剂量、提取时间等。一般情况下,溶剂量越大,有效成分的提取越完全,但是溶剂量过大,产品的能耗增加,因此应选择合适的提取溶剂量,使药物的有效成分刚好能完全提取出。延长提取时间会使有效成分的提取充分,但在有些情况下,长时间高温浸提会增加对提取物破坏的可能性。
2.2.1 提取工艺设计参考有关资料和预实验,以水蒸气蒸馏法提取,选加水倍量(A)、浸泡时间(B)、提取时间(C)作为考察因素,设计3个水平,以挥发油量为考察指标,设计L9(34)正交实验。
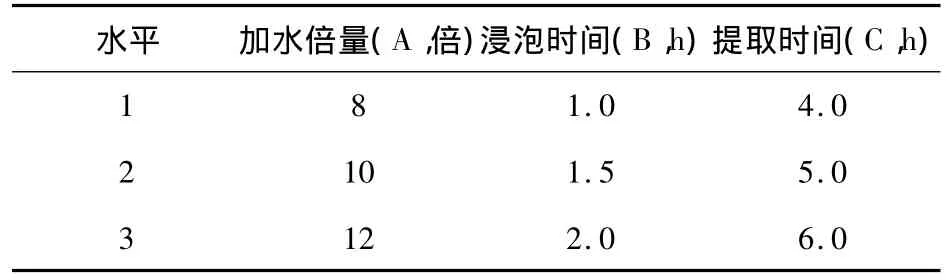
表1 挥发油提取工艺因素水平表
2.2.2 实验方法
根据实验设计,选用L9(34)正交表安排实验。每次实验分别称取除寒水石外其余处方量药材,按正交表各项实验,然后按《中国药典》2005版I部附录挥发油测定法提取挥发油,冷却后称取其量[3]。正交实验结果见表2,方差分析结果见表3

表2 正交实验结果表
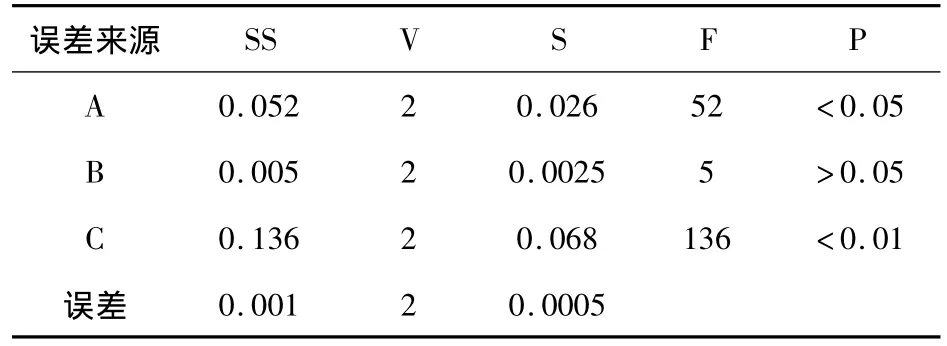
表3 正交实验结果表
以挥发油得量为考察指标,由表2中极差R值大小显示,各因素作用主次为C>A>B>D;表3中方差分析结果表明,C因素的影响具有及显著性意义(P<0.01),A因素的影响具有显著性意义(P<0.05),以A2C2为佳;B因素的影响无显著性意义(P>0.05),因此以A2B1C2组合为佳,即药材加10倍量的水浸泡1h,提取5h。
2.2.3 验证试验
按照A2B1C2最佳工艺方案验证三批,结果见表4。
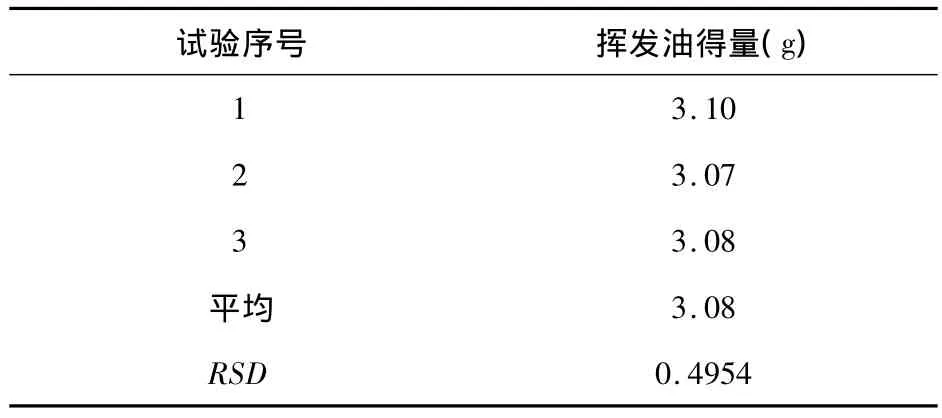
表4 乙醇回流验证试验结果
验证试验结果表明,所筛选的挥发油提取工艺稳定可行。经过不同提取比例的比较,发现最佳提取工艺为药材加10倍量的水浸泡1h,提取5h。
2.3 挥发油包合工艺的筛选和确定:为尽可能地保留挥发油成分,保证疗效,采用β-CD包合挥发油。为了适应生产,采用了操作简便,设备要求不高的饱和水溶液法,对包合时β-CD与油的配比,包合温度及包合时间进行正交实验研究,以β-CD与油的配比(A)、包合温度(B)、包合时间(C)为考察因素,设计三个水平,以挥发油利用率为考察指标,设计L9(34)正交实验,筛选最佳包合工艺条件。
2.3.1 挥发油包合试验因素水平表,见表5。

表5 挥发油包合试验因素水平表
2.3.2 试验方法及结果
按L9(34)正交表安排实验,精密称取 β-CD,置150mL具塞三角瓶中,加入100mL蒸馏水,水浴加热使溶解,降至规定温度,用磁力搅拌器搅拌30min,精密称取挥发油1g,按1:4比例用无水乙醇稀释,用滴管将挥发油稀释液注入β-CD溶液中,加塞,恒温搅拌至规定时间。置冰箱中放置24h,抽滤,用石油醚30mL洗涤三次,置干燥器中12h,即得粉末。将制得的包合物置500mL圆底烧瓶中加入蒸馏水200mL,按挥发油测定法测定包合物中实际含油量;同时进行挥发油空白回收率测定,即将1g挥发油置500ml圆底烧瓶中,按上述方法测定,计算空白回收率;在测定挥发油含量及空白回收率基础上,可计算挥发油利用率,即:挥发油利用率(%)=包合物中实际含油量/挥发油加入量/空白回收率×100%,结果见表6。

表6 正交实验表及结果
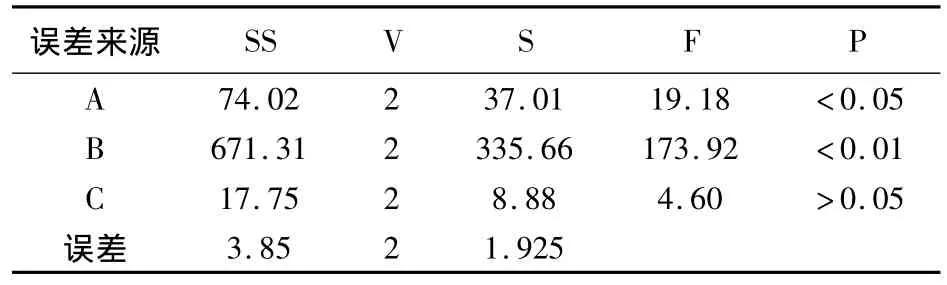
表7 挥发油β-CD包合正交实验方差分析表
挥发油利用率越高,包合效果越好。因此,以挥发油利用率为考察指标,由表6中极差R大小显示,各因素作用主次为B>A>C;方差结果表明:B因素的影响具有极显著意义(P <0.01),A因素影响具有显著性意义(P <0.05),C因素的影响无显著意义(P >0.05),但C2稍优于C1,故以A2 B2 C2组合为佳,即以8倍挥发油量的β-CD包合,包合温度为50℃,包合时间为3h。
2.3.3 验证实验
按最佳包合工艺进行挥发油的包合验证实验,其结果见表8。由表8中结果表明,挥发油的包合工艺稳定可行。
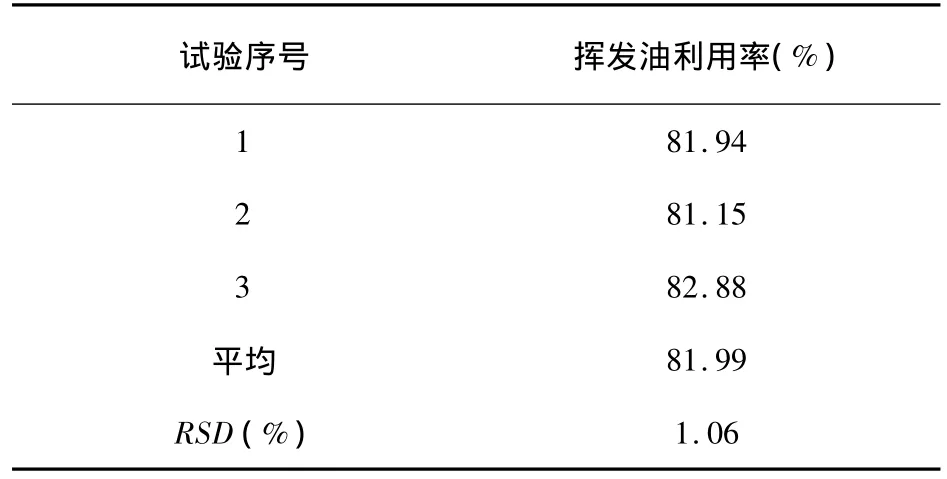
表8 挥发油包合验证实验结果表
2.4 挥发油包合物的定性、定量试验
2.4.1 挥发油包合物薄层定性鉴别试验方法及结果
取挥发油0.15g置100mL量瓶中,加石油醚定容至刻度,作为供试品溶液1;取含0.15g挥发油的β-CD包合物约1.2g,加100mL石油醚洗脱,滤液加石油醚定容至100mL,作为供试品溶液2;继续加100mL石油醚洗脱,滤过,滤液加石油醚定容至100mL,作为供试品溶液3;取石油醚提取后的β-CD包合物加95%乙醇回流提取30min,滤过,滤液加乙醇定容至100mL,作为供试品溶液4;取β-CD1g,加95%乙醇回流提取30min,滤过,滤液加乙醇定容至100mL,作为阴性对照溶液。吸取上述5种溶液各5mL,点于同一硅胶G薄层板上,用石油醚-醋酸乙酯(17:3)为展开剂,展开,取出,凉干,置紫外光灯(365nm)下检视,结果供试品溶液色谱中,在与挥发油溶液相应的位置上,显相同颜色的斑点,而供试品溶液与阴性样品色谱中无此斑点。
2.4.2 挥发油包合物紫外定性鉴别试验方法及结果。
样品1:精密称取挥发油1g,置250mL容量瓶中,加无水乙醇稀释至刻度,摇匀,备用。
样品2:精密称取挥发油包合物9g,用250mL石油醚分5次洗脱(50mL×5),收集洗脱液,低温挥干石油醚,加乙醇溶液备用。
样品3:经石油醚洗脱的挥发油包合物低温干燥,加250mL乙醇回流提取一小时,滤过,收集滤液备用。
样品4:精密称取β-CD 8g,加250mL乙醇回流提取1h,滤过,收集滤液备用。
取上述样品各10mL进行紫外扫描,结果见表9。
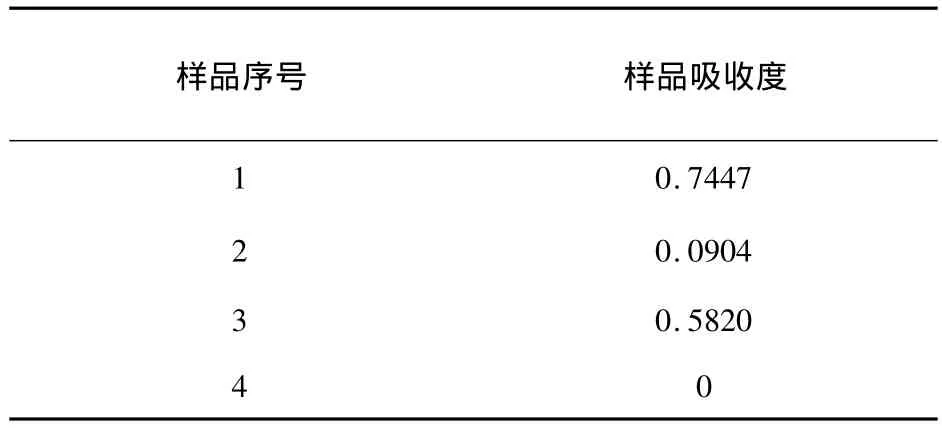
表9 挥发油包合物紫外鉴别试验结果表
由表9中样品吸收度结果表明:2号样品吸收度相对于1、3号样品比较小,说明2号样品是由石油醚洗脱的β-CD表面残留的挥发油,乙醇回流提取已形成包合物的挥发油,故挥发油紫外扫描图与乙醇提取液形成的吸收光谱类似,说明包合物已形成。
综上所述,挥发油的提取,包合工艺相对稳定可行
2.5 水提取工艺研究设计:按照工艺设计要求,对除寒水石外的其余药材进行水提取工艺研究,以溶剂用量(A)、回流时间(B)、回流次数(C)为考察因素进行正交试验,并以浸膏得率为评价指标,考察最佳提取工艺条件。
2.5.1 因素水平表:见表10。
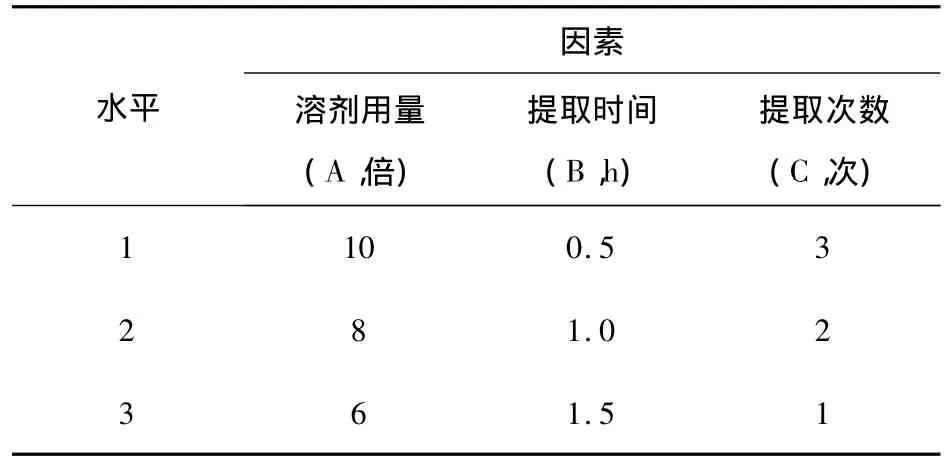
表10 水提取正交试验因素水平表
2.5.2 实验方法
称取处方药材共分9份,按因素水平表安排实验,分别用水进行提取,提取液浓缩,蒸干,称量计算即得。实验设计及结果见表11
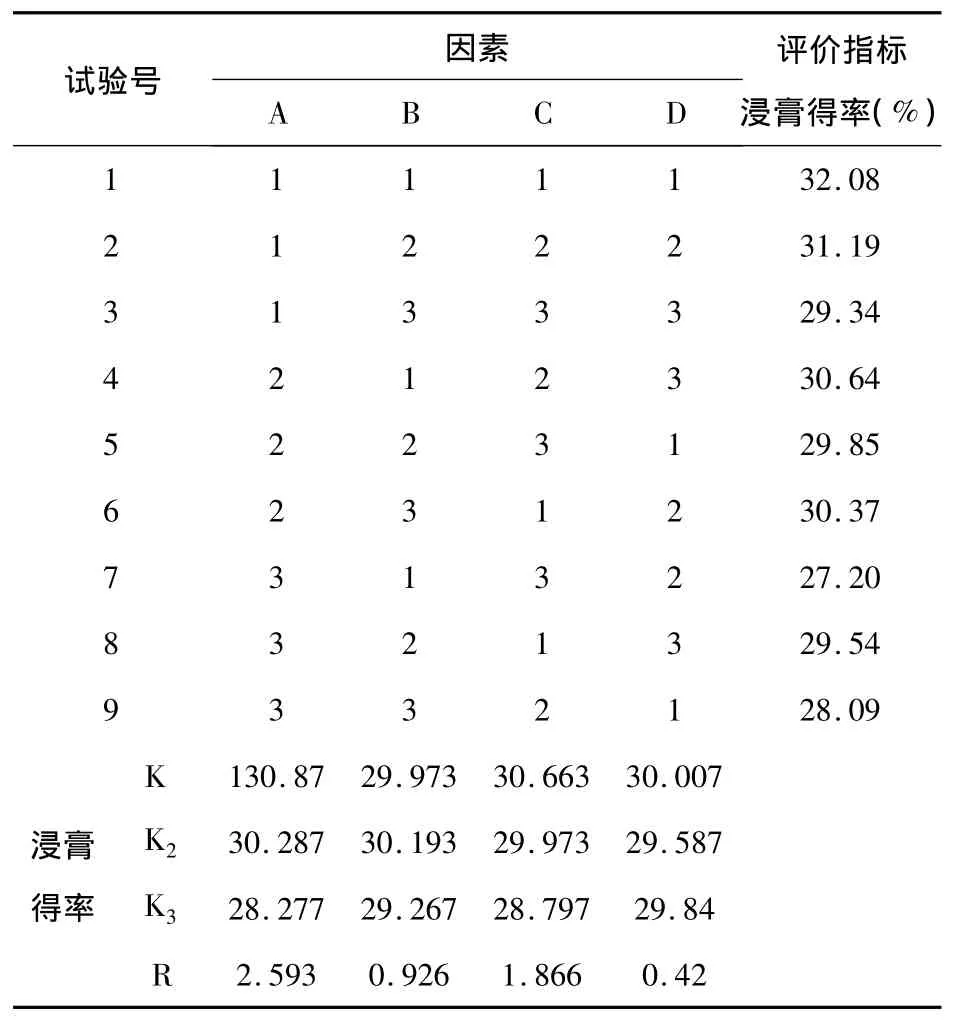
表11 正交实验设计表及结果
2.5.3 将正交实验结果进行方差分析,结果见表12。
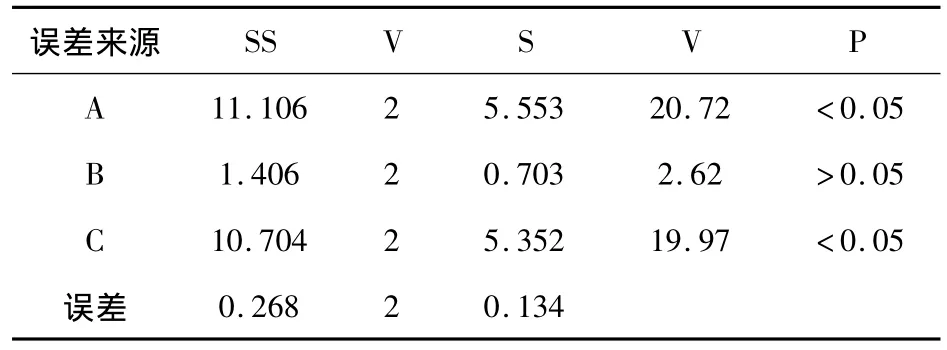
表12 浸膏得率方差分析表
由表11中极差R值大小显示,各因素作用主次为A>C>B,方差分析结果表明,A因素和C因素的影响均具有显著意义(P<0.05),B因素的影响无显著意义(P>0.05),故以A1B2C1组合为佳,即以10倍量水提取1h,共提取3次。
2.5.4 提取工艺验证试验
称取1/9处方量药材按照A1B2 C1进行验证试验,结果见表13。结果表明:正交实验确定的水提取工艺,稳定可行。
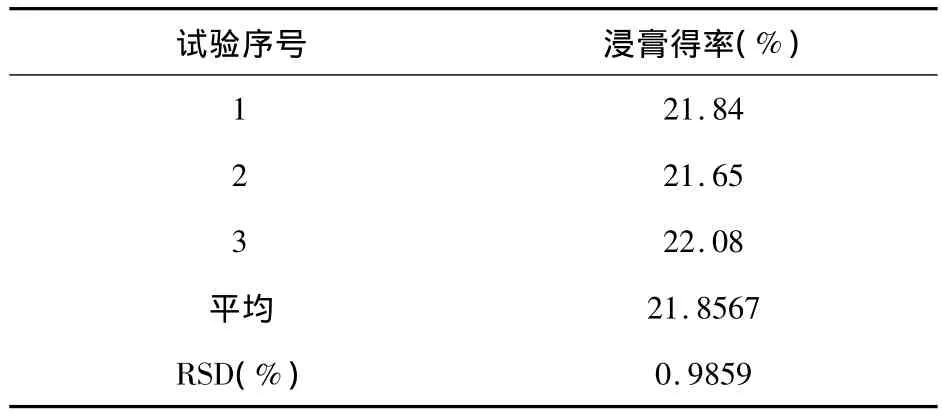
表13 水提工艺的验证试验
2.6 醇处理工艺研究设计:本品处方剂量较大,为减少服用量,保证药物疗效,应将水提膏中混有的淀粉、黏液质等杂质进行纯化处理。结合大生产的生产条件,选用醇沉法,影响醇沉的主要因素有乙醇浓度、水提浓缩液的相对密度、静置时间。以上述三个因素为考察因素,各取3个水平进行L9(34)正交试验。并以醇浸膏得率和阿魏酸保留率为评价指标,筛选最佳醇沉工艺条件。
2.6.1 因素水平表见表14。

表14 醇沉正交试验因素水平表
2.6.2 实验方法
称取处方量药材分为9份,按筛选的最佳水煎工艺条件提取,将提取液浓缩成不同相对密度的浓缩液,按正交试验条件进行醇沉、过滤,回收乙醇,减压干燥。①浸膏得率测定:精密称定干燥物,计算,即得。②阿魏酸保留率测定:精密称取干燥物约0.5g,依“阿魏酸方法”进行含量测定,并计算阿魏酸保留率,即得。③正交实验设计及结果见表15。
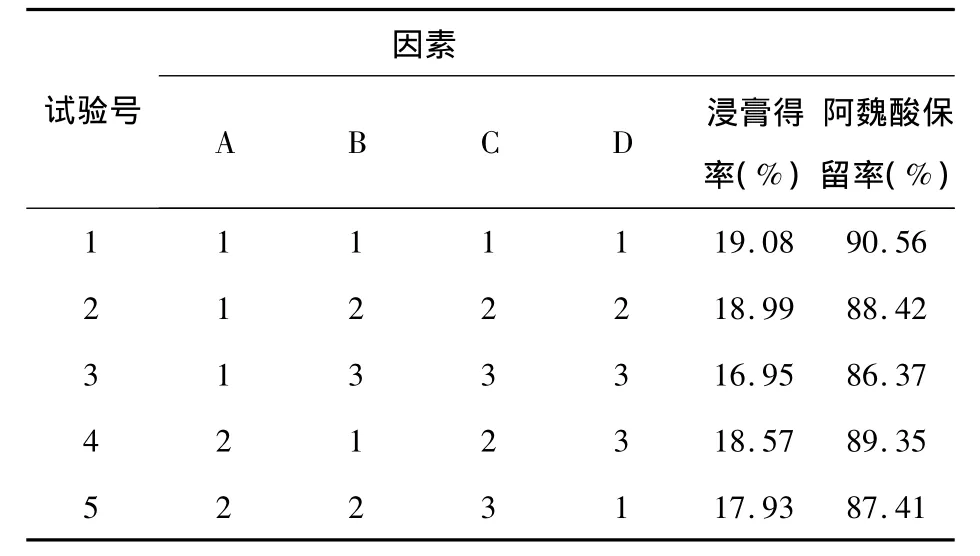
表15 醇沉正交实验设计及结果
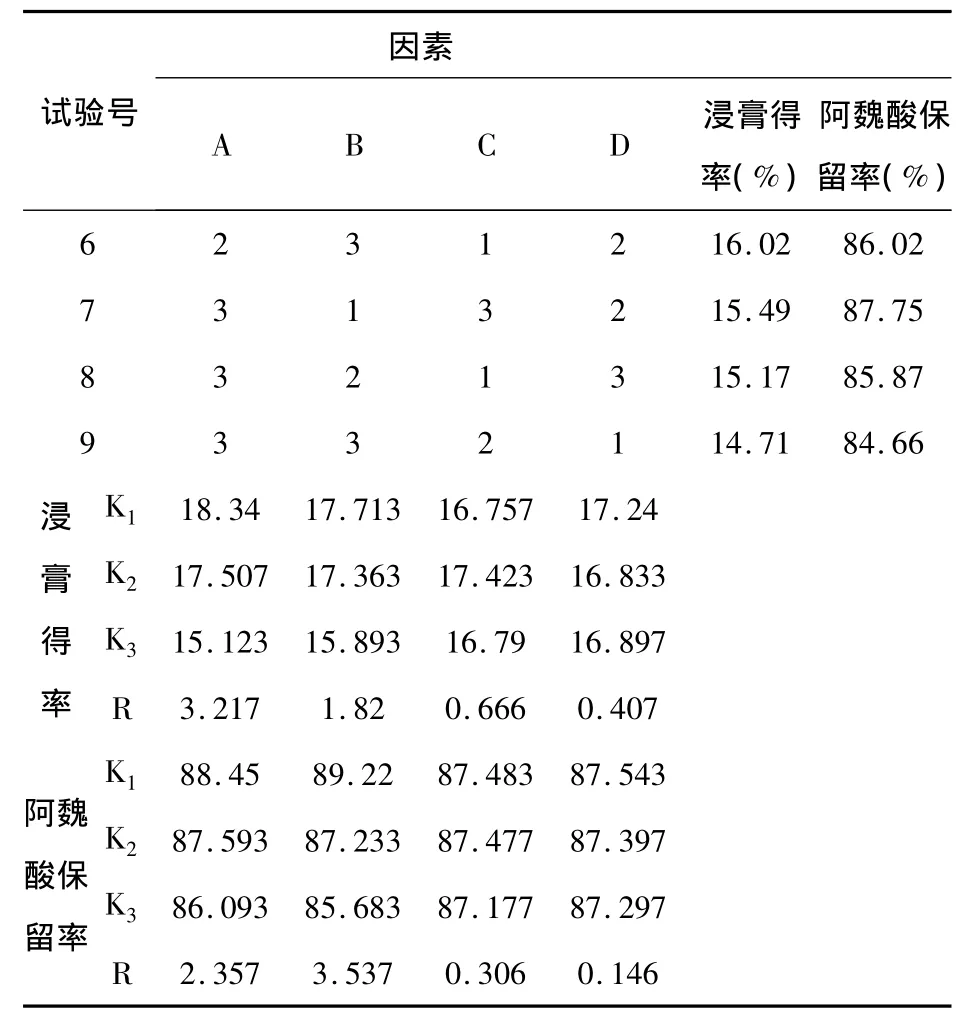
表15 (续)
④将正交实验结果进行方差分析,结果见表16,17。
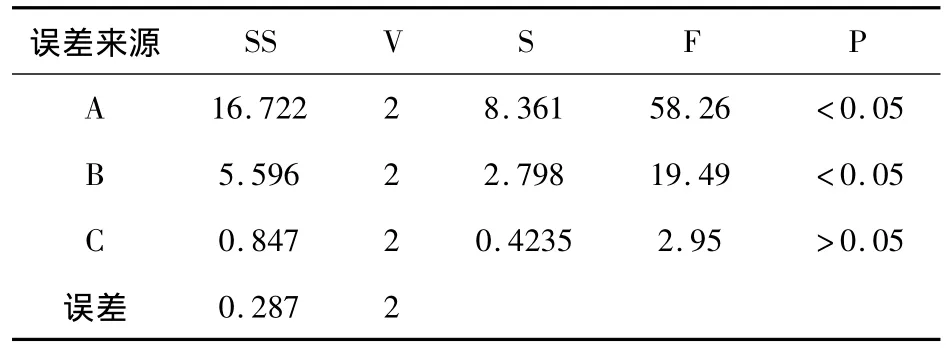
表16 醇沉法浸膏得率方差分析表

表17 阿魏酸保留率方差分析表
以醇沉后浸膏得率为考察指标,由表15中极差R值大小显示,各因素作用主次为A >B>C;方差分析结果表明:A因素和B因素的影响均具有显著性意义(P<0.05),C因素的影响无显著性意义(P>0.05),考虑到应以醇沉后浸膏率少为宜,故以A3B3 C2组合为佳;以阿魏酸保留率为考察指标,由表15中极差R值大小显示,各因素作用主次为B>A>C;方差分析结果表明:A因素和B因素的影响均具有显著性意义(P<0.05),C因素的影响无显著性意义(P>0.05),故以A2B3 C1组合为佳;考虑到阿魏酸保留率指标的重要性,故以A2B3 C1为最佳醇处理工艺。即药液浓缩至相对密度为1.30,使药液含65%乙醇,静置24h。
2.6.3 验证试验
按照A2B3C1最佳工艺方案验证三批,结果见表18。验证试验结果表明,所筛选的提取工艺稳定可行。

表18 醇处理验证试验结果
2.7 成型工艺研究
2.7.1 实验前准备:将醇提膏干燥,粉碎,过120筛,与过200目筛的寒水石及挥发油包合物混和,备用。一个处方混合物大约为318g。
2.7.2 药物与基质配比的初选实验设计:为了尽量减少基质的用量,以减小服用量,同时也为后续研究工作提供依据,根据药物性质选择基质分别为PEG4000和PEG6000,药物与基质的配比分别为 1:1.5,1:2.0,1:2.5,1:3.0,1:3.5,1:4.0进行滴制试验。以滴制难易程度和滴丸圆整度为依据进行筛选,实验结果见表19。根据实验结果可知,药物与基质的比例最佳为1:2.5。

表19 药物与基质配比的初选
2.7.3 复合基质配比的选择
按药物与基质配比为 1:2.5,考察 PEG4000与PEG6000 按2:0.5,2:1,2:2,2:3,2:4,2:5 的配比进行滴制试验。以药液黏度、滴丸圆整度与溶散时限为依据进行筛选,实验结果见表20。根据实验结果可知,复合基质比例最佳为2:1。

表20 复合基质配比的选择
2.7.4 冷却剂的选择:根据确定的药物与基质的配比,进行预试验,以液体石蜡为冷却剂能满足滴丸剂的需求,因此确定滴丸冷却剂为液体石蜡。
2.7.5 滴制工艺实验研究:在单因素筛选的基础上,以基质种类、药物与基质配比、药液温度、冷却剂温度作为考察因素,各取3个水平进行L9(34)正交试验。并以丸重变异系数,溶散时限和外观质量为评价指标,筛选最佳滴制工艺条件,因素水平见表21。
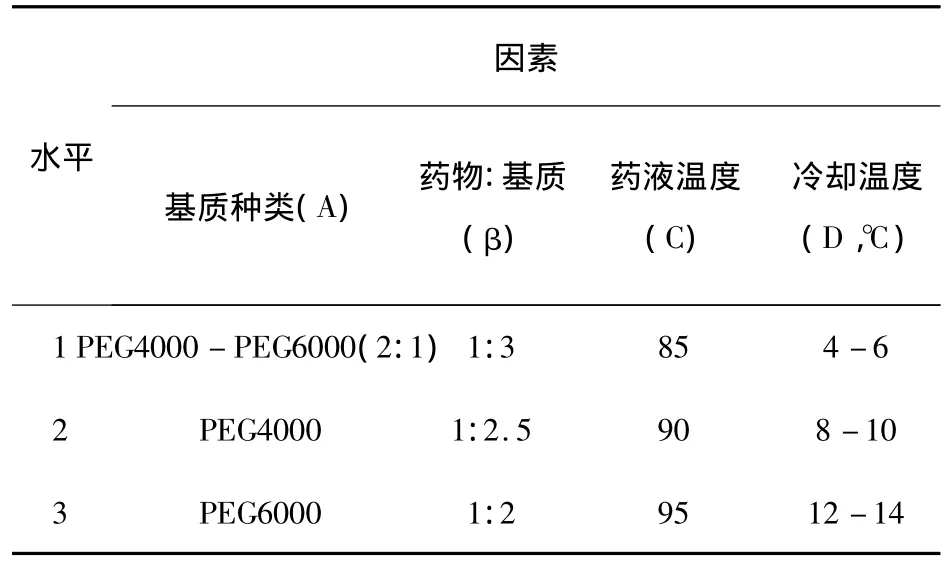
表21 滴制工艺筛选因素水平表
①正交实验方法及结果:按照L9(34)正交表安排实验。将基质置水浴中加热搅拌至全部熔融后,加入规定比例的药物,搅匀,迅速移至已预热至恒温的滴丸机滴料罐中,保持规定温度,将药液滴入液体石蜡冷却剂中。取出滴丸,吸除表面液体石蜡,于干燥器中放置24 h,即得实验样品。每次取实验样品按《中国药典》(2005年版)方法测定溶散时限,然后随机抽取20粒,精密称定总重量,再分别精密称定各丸的重量,求出平均丸重的变异系数,并用10分制对包括滴丸成型性、外形、圆整度、硬度和色泽均匀度在内的外观质量评分。结果见表22

表22 滴制工艺正交实验安排及实验结果
②实验结果分析:滴丸的质量不能仅用一个指标衡量,本实验采用3个评价指标进行数据处理,求得不同指标下各因素的极差(R),从R值大小判定影响因素的主次。若选用各因素不同水平下,滴丸丸重变异系数最小,溶散时限最短,外观质量分最高为最佳搭配 ,则3个指标下可供选择的最佳工艺条件,分别归纳于表23中。
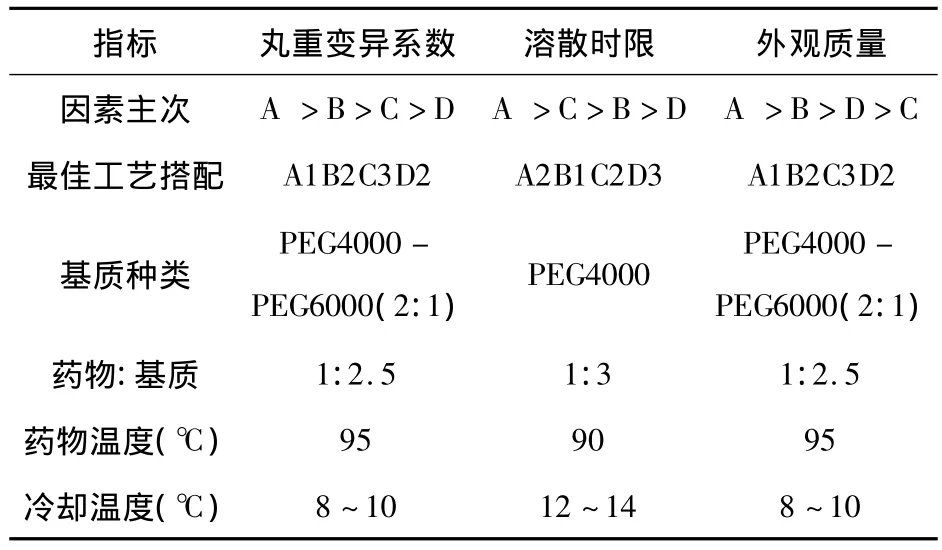
表23 正交实验分析结果表
综合表23中各指标下最佳工艺搭配,选择两个或两个以上指标中均为较佳的工艺条件,得优选工艺条件为A1B2C3D2,即以PEG4000-PEG6000(2:1)为基质,药物与基质配比为 1:2.5,药液温度为95℃,冷却剂温度为 8~10℃。
③验证实验:以上述确定的最佳工艺条件重复制备3批滴丸样品,并按《中国药典》2005年版制剂通则滴丸剂项下进行质量检查。结果见表24。
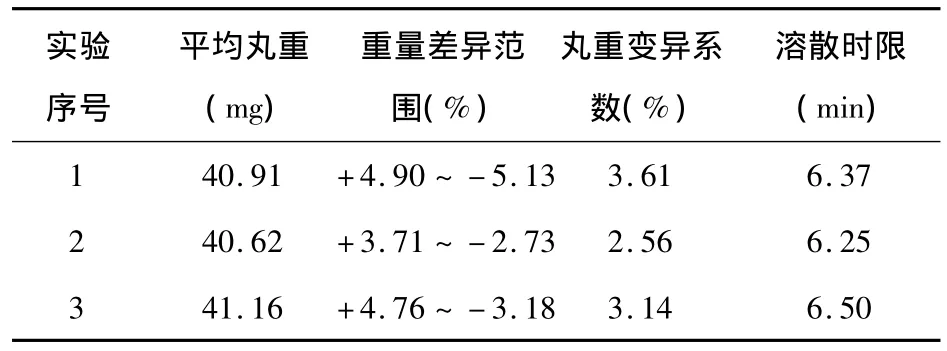
表24 滴丸滴制工艺验证实验结果表
验证结果表明:验证结果与正交实验结果相近,说明滴丸制备工艺基本稳定可行。
3 愈心安神滴丸制备工艺总结
六味药中,除寒水石外,其余药材提取挥发油,用β-环糊精包合,备用。药渣加水煎煮3次,滤过,合并滤液,滤液浓缩成相对密度为1.26~1.28(50℃)的清膏,加乙醇使含醇量达65%,搅匀,静置24h,取上清液,回收乙醇,浓缩干燥,粉碎并过120目筛,备用,寒水石研细,过200目筛。取聚乙二醇4000与聚乙二醇6000适量加热使溶解,加入上述挥发油包合物,稠膏(干)和寒水石研细,混匀,滴入冷却的液体石蜡中,制成滴丸,即得。
[1]谢秀琼.中药新制剂开发与应用[M].人民卫生出版社,1994.
[2]杨基森,等.中药制剂设计学[M].贵州科技出版社,1992.
[3]中华人民共和国国家药典委员会.中国药典一部[M].北京:化学工业出版社,2005.
猜你喜欢
纺织科技进展(2021年3期)2021-06-09
天津中医药(2020年5期)2020-06-01
中央民族大学学报(自然科学版)(2018年1期)2018-06-27
天然产物研究与开发(2018年10期)2018-03-08
中成药(2017年12期)2018-01-19
中成药(2017年7期)2017-11-22
中成药(2017年10期)2017-11-16
中成药(2017年5期)2017-06-13
药学研究(2015年11期)2015-12-19
中药与临床(2015年5期)2015-12-17
