
季戊四醇双缩醛水解反应过程的1H NMR在线研究
2013-11-28 00:59张国君孙小强李正义周蓓蓓
分析测试学报 2013年1期
张国君,杨 扬,孙小强,李正义,周蓓蓓,殷 乐
(常州大学 江苏省精细化工重点实验室,江苏 常州 213164)
季戊四醇双缩醛(酮)类物质用途广泛。工业上常用作杀虫剂、增塑剂、抗氧化剂和表面活性剂的消泡剂等[1-5];在有机合成中常作为潜在的保护基团合成具有生物活性和药物传输功能的物质[6];也可用作医用外科手术缝线、药物缓释、骨科固定、组织修复等材料[7-8]和具有特殊功能的新型包装材料[9]。近年来,可降解材料已成为材料领域的研究热点[10]。由季戊四醇双缩醛类化合物参与合成的聚合物由于含有多个易水解的醚键,而具有易降解性能。为此,通过研究季戊四醇双缩醛类化合物的水解反应历程,可有效地控制反应,为此类化合物的绿色水解提供基础数据。
核磁共振(NMR)技术作为一种日益成熟的仪器分析方法,具有传统方法不可比拟的优势。动力学核磁共振是核磁共振波谱学中有一定独立性的一个分支,它以NMR为工具,研究某些化学动力学过程,如分子结构和构象,蛋白质及其配位的相互作用;化学交换及立体化学反应速度及其他动力学问题,从而得到动力学和热力学参数[11-12]。孙先勇等[13]利用原位的29Si液体核磁,研究了氨水催化条件下TEOS/DDS混合体系原位共水解的动力学过程;顾开春等[14]利用核磁共振方法研究了Michael加成反应的动力学行为及反应机理;另外,在结构基因组学和机构生物学的研究中,核磁共振技术被广泛地用于测定蛋白质在溶液中的空间结构及其动力学,研究蛋白质与配体的相互作用[15-16]。本实验在核磁管中进行,采用1H NMR技术跟踪研究水解反应历程,具有所需样品含量少、速度快、重复性好、不终止反应等优点。

图1 物质A、B、C、D、E、F、G、H和I的结构式Fig.1 Structure diagrams of compound
1 实验部分
1.1 试剂与仪器
3,9-二-(4-甲氧基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(4-氨基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-苯基-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(4-氯苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(4-硝基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(3-氨基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(2-氨基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(3-硝基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷、3,9-二-(2-硝基-苯基)-2,4,8,10-四氧杂螺[5.5]十一烷等季戊四醇螺环化合物由本实验室制备,纯度均在98%以上。下文分别以物质A、B、C、D、E、F、G、H和 I表示以上9种化合物,其结构式见图1。DMSO-D6(CIL公司,氘代度为 99.9%)、D2O(SAFC公司,氘代度为99.9%)、盐酸(国药集团化学试剂有限公司,分析纯)。
所有1H NMR实验均在 Bruker AVANCEⅢ400超导NMR仪上完成。实验参数:1H NMR观测频率为400.13 MHz,5 mm BBO探头,zg 30单脉冲序列,谱宽8 012.82 Hz,中心频率2 600.84 Hz,扫描8次,弛豫时间1 s。
1.2 实验方法
准确称量0.001 0 g的水解反应物于外径为5 mm的核磁管中,加入0.4 mL DMSO-D6使其完全溶解,再加入0.1 mL用D2O稀释的盐酸(稀释后pH值分别为0、1、2、3、4),放置于预先调好温度的超级恒温器中水解。采用核磁管内置毛细管的方法进行定量分析,密封毛细管内加入65 μL氘代水和15 μL丙酮的混合物。根据反应速度每隔一段时间对其进行检测。以DMSO的质子残存峰为化学位移基准,以丙酮峰面积为积分基准,对所获取的谱图进行处理,获取相关谱线的相对积分值,以此为依据对水解过程中反应体系内各物质的浓度变化情况进行分析。
2 结果与讨论
2.1 溶剂的选择
该反应为水解反应,水作为一种反应物参与到反应体系中,因此选择能与水互溶的溶剂将有利于反应的进行。经实验证实,在与水互溶的甲醇、乙醇、四氢呋喃、乙腈和二甲基亚砜中,反应的效果较好,且与水的互溶性越好,反应速率越快。由于该反应在核磁管中进行,并以1H NMR技术对反应进行全程跟踪,考虑到季戊四醇双缩醛类物质自身的溶解性和溶剂与水的互溶性,本实验选用氘代DMSO作为溶剂。
2.2 初始浓度对水解反应的影响
水解初始浓度对反应存在一定影响。本实验中当初始浓度过高时,随着酸的加入,会有不同程度的反应物析出;当初始浓度过低时,反应物的信号峰又极其微弱;所以初始浓度过高或过低均不利于观察反应,不能准确反映各物质峰面积的变化。综合考虑以上因素,反应物的初始浓度选定为2.0 g·L-1。
2.3 水解反应机理
在酸性条件下季戊四醇双缩醛的水解反应为羟醛缩合的逆反应,水解缩醛的实质是水与羰基碳的亲核加成脱去醇。氢质子首先进攻螺环上的缩醛氧原子,然后质子化的氧原子发生碳氧键的断裂。带正电荷的氧原子吸引电子的程度加强,使得羰基碳的正电性提高,水分子对羰基碳进行亲核加成,然后质子化的氧原子发生碳氧键断裂,再去质子化,得到产物。下面以物质A为例,其反应机理见图2。

图2 物质A的水解反应机理Fig.2 Hydrolysis mechanism of compound A
2.4 1H NMR水解谱图分析
以物质A为例,考察了其在不同时刻的水解核磁谱图(图3)。图3a为物质A水解0 min的核磁谱图,δ7.35、δ6.91为苯环上 H4(4')、H5(5')(见图1)的质子共振信号;δ5.43为与杂原子氧相连叔碳上H3(3')的质子共振信号;δ4.54、δ3.87、δ3.77、δ3.64分别为螺环化合物亚甲基上 H1e(1'e)、H1a(1'a)、H2e(2'e)、H2a(2'a)的质子共振信号;δ3.75为甲氧基上H6(6')的质子共振信号,且与其中的一组亚甲基峰相互重叠;δ4.23和δ1.71分别为内标中氘代水和丙酮的质子共振信号;δ3.34为DMSO-D6中水的质子共振信号,δ2.50为DMSO-D6的质子残存峰。

图3 物质A在不同时刻的水解核磁谱图(400 MHz,DMSO-D6)Fig.3 NMR spectra of compound A during different hydrolysis time hydrolysis time(a-e):0,30,90,150,240 min
图3b为物质A水解30 min后的核磁谱图,随着酸的加入,水解反应的进行伴随着新物质的生成并产生了对应的信号峰,整个反应体系内的质子共振信号发生变化。δ9.80为水解产物对甲氧基苯甲醛对应的醛基质子共振信号,δ7.85、δ7.10为对甲氧基苯甲醛苯环上质子的共振信号;δ5.31为中间产物单缩醛的一个特征质子共振信号;甲氧基上的质子共振信号也发生了细微变化,由δ3.75移至δ3.82;δ3.36为水解产物季戊四醇亚甲基上质子的共振信号;内标中氘代水和丙酮的质子共振信号分别移至δ4.29和δ1.80处;δ3.92为加入的酸中氘代水的质子共振信号;由于使用的盐酸已用D2O稀释,季戊四醇羟基氢被D2O交换,所以观察不到羟基的质子共振信号。其它质子共振信号的归属与图3a相同。
图3中c、d的变化情况类似于图3b,但随着反应的继续进行,反应物信号峰峰面积越来越小,直至完全消失,中间产物的信号峰峰面积先增加后下降,直至水解为最终产物,最终产物的信号峰峰面积则持续增加,直至水解反应彻底结束。
图3e为物质A最终水解时的核磁谱图,δ9.80为最终水解产物对甲氧基苯甲醛醛基的质子共振信号;δ7.85和δ7.10为对甲氧基苯甲醛苯环上的质子共振信号;δ3.82处为对甲氧基苯甲醛甲氧基上的质子共振信号;δ3.36为季戊四醇亚甲基上的质子共振信号;δ3.92为加入的酸中氘代水的质子共振信号;δ4.29和δ1.80为内标中氘代水和丙酮的质子共振信号;δ2.50为DMSO-D6的质子残存峰。综上所述,核磁谱图能够观察反应体系中各组分的含量变化,并能定性和定量分析水解反应的全过程及最终水解产物,所以1H NMR能跟踪监测季戊四醇双缩醛水解反应。
2.5 水解重现性实验
在温度为323.15 K、pH=2.0、物质A初始浓度为2.0 g·L-1的条件下进行3次重现性实验,结果如图4所示。从图4可以看出,实验数据呈现良好的重现性。
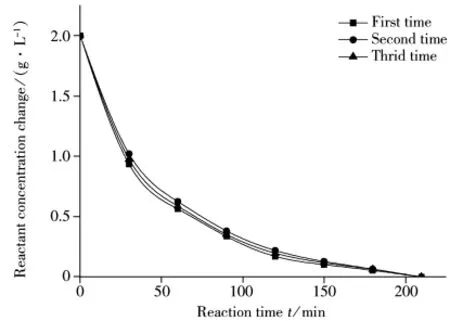
图4 物质A水解重现性实验Fig.4 Repeatability tests for compound A hydrolysis
2.6 反应中各组分的含量变化
酸性条件下,季戊四醇双缩醛的水解反应为连串反应,反应物双缩醛的含量随着反应时间的延长而逐渐降低。反应过程中产生的中间产物单缩醛,在反应初期的含量为0;随着反应的进行,其含量逐渐增加,在某一时刻含量达到最高,但随着反应的继续进行,中间产物会进一步水解,直至水解为最终产物季戊四醇和对应的苯甲醛。图5为物质A在333.15 K、pH=2.0条件下水解过程中各物质的含量变化图。
2.7 反应速率常数的计算
由核磁图谱给出相对积分面积,由计算公式转化为相应的浓度,计算公式如下:

式(1)中Ai、As和A0分别为组分i、内标物和起始原料的峰面积;Ci和C0分别为组分i和起始原料的浓度。
假设季戊四醇双缩醛的水解反应为一级反应,则有:

图5 物质A水解过程中各物质的浓度变化Fig.5 Concentration variation of different components during hydrolysis of compound A

式中,CA表示反应至某一时刻t时反应物的浓度;t表示时间;k表示反应速率常数;C表示常数。实验测得一系列不同时刻的反应物的相对峰面积,并通过公式(1)计算出相应的浓度CA,用Origin作lnCA~t图,呈直线关系,可确定假设成立。所以此类季戊四醇双缩醛的水解反应为一级反应,该直线斜率即为反应速率常数k。
2.8 温度与pH值对水解反应速率的影响
综合考虑以上各种季戊四醇双缩醛的水解速率,选择物质A为研究对象,考察其在不同反应温度和不同酸性条件下的水解情况。
由图6可知,在一定范围内,物质A的水解速率随温度的升高、酸性的增强而加快。从分子运动理论和碰撞理论来看,温度升高后,季戊四醇缩醛的分子动能增加,运动加快,与溶液中H+碰撞的几率增加,被质子化的几率和速率增加。初始浓度和温度相同的条件下,在一定范围内,酸性越强越利于水解。在反应过程中氢质子进攻缩醛氧原子为反应速率的控制步骤,酸性越强越有利于质子化,所以pH值对水解反应有极其重要的影响。通过改变以上两种条件,可在工业水解时寻求较佳的水解条件来减少能源和资源的消耗。

图6 物质A在不同温度,不同pH条件下水解速率变化趋势Fig.6 Hydrolysis rate changing trends of compound A under different temperature and different pH
2.9 苯环上不同取代基对水解速率的影响
为了探究苯环上不同取代基对水解速率的影响,分别考察了物质A、B、C、D和E在不同温度、不同酸性条件下的水解速率,结果如图7所示。

图7 不同取代基季戊四醇双缩醛的水解速率变化趋势Fig.7 Hydrolysis rate changing trends of different pentaerythritol dialdehydes compound E could not be hydrolysed under these conditions
研究表明,物质A和B在酸性较强时极易水解,无法及时检测,因此实验选择在相对弱酸(pH 4.0)条件下对A和B进行水解。而在此条件下C、D、E基本不发生反应,这表明A、B比C、D、E更易进行水解。从图7中可知,B水解趋势曲线明显高于A,因此在相同条件下,B的水解速率更快,比A更易进行水解。同时,实验选取酸性较强(pH 1.0)条件对C、D、E进行水解。从图7中C、D的水解趋势曲线可知,在同一温度、pH值条件下,C的水解速率常数大于D,这表明C比D更易水解。而E在此条件下不发生水解反应,所以上述5种化合物水解从易到难分别为物质B>A>C>D>E。这主要是由苯环上取代基不同而造成的。反应过程中氢质子进攻缩醛氧原子为反应速率的控制步骤,其水解速率依赖于底物反应中心上的电子云密度,高的电子云密度有利于亲电试剂的进攻,低的电子云密度有利于亲核试剂的进攻。物质A和B苯环上的取代基是推电子基团,使苯环上的电子云密度升高,有利于质子化,且—NH2的推电子能力比—OCH3强,所以B比A更易水解;物质D和E苯环上的取代基是吸电子基团,使苯环上电子云密度降低,不利于质子化,且—NO2的吸电子能力比—Cl强,所以E比D更难水解。综上所述,苯环上取代基对水解的难易程度有着非常重要的影响。
2.10 苯环上取代基位置对水解速率的影响
为了探究苯环上不同取代基位置对水解速率的影响,分别测定了胺类取代和硝基类取代季戊四醇双缩醛的水解速率,结果如表1所示。
由表1可知,在相同温度(313.15 K)和酸性条件下(pH 4.0),氨类季戊四醇双缩醛F(间位)不发生水解,B(对位)的水解速率常数约是G(邻位)的10倍,这表明B最易水解,G次之,F最难水解。在相同温度(333.15 K)和相同酸性条件下(pH=0),硝基类季戊四醇双缩醛H(间位)、E(对位)和I(邻位)的水解速率常数分别为0.001 4、0.000 6、0.000 2,这表明H最易水解,E次之,I最难水解。由此证明,同一取代基在苯环上的取代位置不同,对水解速率会产生较大影响,而且对于吸电子基和推电子基不能一概而论。对于推电子基而言,它使所连苯环上的电子云密度增加,尤其在邻、对位处增加得更多,对苯环有活化作用,有利于亲电试剂的进攻,但邻位相对于对位的空间位阻较大,不利于H+进攻缩醛氧原子,所以对于推电子基的物质,水解速率由快到慢依次为对位>邻位>间位。对于吸电子基团而言,它使所连苯环上的电子云密度降低,并且在邻、对位处降低得更多,对苯环有致钝作用,不利于亲电试剂的进攻,但相比之下,间位的电子云密度较高,亲电试剂进攻间位较为有利,所以推电子基水解速率由快到慢依次为间位>对位>邻位。

表1 胺类与硝基类季戊四醇双缩醛苯环上不同取代位置的水解速率常数Table 1 Hydrolysis rate constant of different positions of substituted groups of the amino and nitro on the aromatic ring of pentaerythritol dialdehyde
3 结论
本文建立了一种在核磁管中考察季戊四醇双缩醛水解反应的新方法。通过对季戊四醇双缩醛水解速率的测定可知,此类化合物的水解速率随温度的升高、酸性的增强而加快。同时,本文还研究了苯环上取代基类型和位置对此类化合物水解速率的影响。研究结果表明,苯环上推电子基团有利于水解反应,同一推电子基水解速率由快到慢依次为对位>邻位>间位;吸电子基团不利于水解反应,同一吸电子基水解速率由快到慢依次为间位>对位>邻位。此方法具有灵敏度高,不破坏样品和节约原料等特点,符合绿色环保的要求。但该方法对反应速度极快和溶解性差的化合物存在一定的局限性。
[1]Yuan X Y,Zhang M,Wang X Y.Chem.Reagents(袁先友,张敏,王小勇.化学试剂),2006,28(9):541-543.
[2]Wang M,Song Z G,Jiang H,Gong H.Chem.Res.Appl.(王敏,宋志国,姜恒,宫红.化学研究与应用),2008,20(1):75-79.
[3]Liu Q F,Wang C P,Zhang J,Tan T F,Zhai X L.J.Hebei Normal Univ.:Nat.Sci.Ed.(刘清福,王春平,张杰,谈廷凤,翟学良.河北师范大学学报:自然科学版),2005,29(6):595-597.
[4]Zhang Z H,Li T S,Jin T S,Li J T.J.Chem.Res.(S),1998,(10):640 -641.
[5]Wang G W,Yuan X Y ,Liu Y C ,Lei X G.J.AOCS,1994,71(7):727-730.
[6]Barr J,Woodburn K W,Ng S Y,Shen H R,Heller J.Adv.Drug Delivery Rev.,2002,54(7):1041 -1048.
[7]Zhao Y M,Wang Z Y,Yang F.J.Appl.Polym.Sci.,2005,97(1):195 -200.
[8]Kricheldorf H R.Chemosphere,2001,43(1):49 -54.
[9]Kim K S,Lee S M,Ryu K C,Lee K S.Polym.Bull.,1995,35:57 -63.
[10]Ge J J.Biodegradable Polymers and Its Application.Beijing:Chemical Industry Press(戈进杰.生物降解高分子材料及其应用.北京:化学工业出版社),2002.
[11]Dai Y.J.Anhui Univ.:Nat.Sci.Ed.(戴怡.安徽大学学报:自然科学版),1998,22(2):89-94.
[12]Foster M P,McElory C A,Amero C D.Biochemistry,2007,46(2):331-340.
[13]Sun X Y,Xu Y,Xu W J,Wu D,Sun Y H,Yang Y X,Yuan H Z,Deng F.Acta Chim.Sin.(孙先勇,徐耀,徐武军,吴东,孙予罕,杨永霞,袁汉珍,邓风.化学学报),2005,63(23):2103-2111.
[14]Gu K C,Yang G,Zhang W P,Liu X M,Yu Z S,Han X W,Bao X H.Chin.J.Inorg.Chem.(顾开春,杨刚,张维萍,刘秀梅,余正坤,韩秀文,包信和.无机化学学报),2006,22(6):1044-1048.
[15]Shi Y H,Guo C Y,Lin D H.Chem.Life(施燕红,郭晨云,林东海.生命的化学),2006,26(2):166-168.
[16]Goto N K,Kay L E.Curr.Opin.Struct.Biol.,2000,10(5):585 -592.
猜你喜欢
中学化学(2022年5期)2022-06-17
大学化学(2022年1期)2022-02-28
合成树脂及塑料(2020年1期)2020-03-27
高中数理化(2020年1期)2020-02-29
四川师范大学学报(自然科学版)(2018年2期)2018-04-28
化工管理(2017年12期)2017-03-04
当代化工研究(2016年7期)2016-03-20
中国塑料(2015年10期)2015-10-14
中国塑料(2015年7期)2015-10-14
昌吉学院学报(2013年1期)2013-12-08
