
EPO对AD样大鼠海马组织氧化应激反应的影响
2013-11-25 10:47李宜培
河南医学高等专科学校学报 2013年2期
靳 力,李宜培,王 黎△
(1.郑州大学基础医学院,郑州450052;2.河南卫生职工学院,郑州451191)
阿尔茨海默病(Alzheimer’s disease,AD)是一种与衰老相关的神经退行性疾病,其组织病理学特征表现是老年斑的沉积,神经纤维的缠结,神经元和突触的丢失[1]。其中淀粉样蛋白-β 是老年斑的主要成分,并且在阿尔茨海默病的发生发展过程中扮演了重要的角色,尽管Aβ 引起AD 发病的确切机制仍不清楚,但许多证据提示由Aβ 激起的过强的氧化应激反应是AD 一个重要的致病因素[2-3]。此外,在Aβ 诱导的神经退行性疾病的研究中发现,过多活性氧自由基的产生能够导致神经元细胞的凋亡[4-5]。所以,抗氧化治疗是AD 病治疗的重要策略之一。
EPO 是一种调控红细胞生成的糖蛋白,临床上被广泛的引用于治疗各种贫血症状。近年来发现,EPO 在多种脑损伤和神经系统病变中发挥重要的神经保护作用[6-10]。并且,相关研究发现EPO 具有对抗氧化应激的功能[11-12]。因此,本实验应用Aβ1-42复制AD 大鼠模型,观察EPO 对AD 大鼠海马总抗氧化能力、过氧化氢酶、一氧化氮、一氧化氮合成酶含量的影响,并探讨其作用机制。
1 材料与方法
1.1 材料 雄性SD 大鼠40 只,清洁级,体质量220 ~250 g,动物批号:SCXK(豫)2010 ~0002,由河南省实验动物中心提供。经Y 迷宫筛选淘汰4 只后,其余随机分为3组:生理盐水组,AD 模型组和EPO 治疗组,每组12 只。自由饮食,昼夜节律,室温保持在20 ℃。
1.2 主要试剂和仪器 总抗氧化能力测试盒,过氧化氢酶测试盒,一氧化氮测试盒,一氧化氮合成酶测试盒以上试剂均购自南京建成生物工程研究所;可见光分光光度计(北京普系统用仪器责任有限公司);组织匀浆器(宁波新芝生物科技股份有限公司);超速冷冻离心机(德国backman 公司)。
1.3 Aβ1-42孵育及模型制作 将Aβ1-42溶于无菌生理盐水中,稀释成10 ug/ul 的浓度,置于37 ℃恒温箱孵育一周,使其变为聚合态。用6%水合氯醛(30 mg/kg)对大鼠进行腹腔注射麻醉,将麻醉后的大鼠头顶部备皮并固定于脑立体定位仪上,以前囟为定位原点,参照《大鼠脑立体定位图谱》定位大鼠海马CA1 区:前囟后4.0 mm,中线旁2.0 mm,颅骨下进针3.6 mm。用微量进样器对每只大鼠双侧海马进行Aβ1-42注射:每只每侧注射1 ul,注射10 min,留针10 min,以确保溶液充分弥散,随后缓慢撤针,骨蜡封堵针孔,缝合消毒皮肤,术后每只肌注10 万单位青霉素预防感染。
AD 模型组和EPO 治疗组大鼠海马注射Aβ1-42,生理盐水组大鼠海马注射等量生理盐水。EPO组从造模当天起腹腔注射rHu -EPO,剂量为5 000 IU/kg,隔天一次,共5 次。
2 检测方法
2.1 测试样品的制备 取海马置于4 ℃的生理盐水中漂洗,去除血液,滤纸拭干,称重,按重量∶体积=1∶9 的比例加入4 ℃的生理盐水,用组织匀浆器充分匀浆,将匀浆液以2 500 转/分,离心10 ~15 min,取上清液,并用用考马斯亮蓝法测定总蛋白含量,-80 ℃冻存备用。取已处理好的待测样品上清液于4 ℃冰箱解冻,浓度为10%。严格按照实验操作表进行工作液的配制和对样品的检测。测定海马组织总抗氧化能力及过氧化氢酶、一氧化氮、一氧化氮合成酶的含量。
2.2 统计学处理 数据采用单因素方差分析,应用SPSS19.0 软件系统进行统计学处理,组间比较用t检验,a=0.05 作为检验标准。
3 结果
3.1 各组大鼠海马匀浆总抗氧化能力的变化 结果如图1 所示:与生理盐水组相比较,AD 模型组的总抗氧化能力明显地下降(P <0.05),而EPO 治疗组与AD 模型组相比总抗氧化能力显著地升高(P <0.05),差异具有统计学的意义。

表1 各组大鼠T-AOC 能力,CAT 活力,NO 含量和NOS 活性的变化(n=12)
3.2 各组大鼠过氧化氢酶活性的变化 结果如图2 所示:与生理盐水组相比较,AD 模型组的过氧化氢酶活性明显地下降(P <0.05),同时,EPO 治疗组与AD 模型组相比,过氧化氢酶活性显著地升高(P<0.05),差异具有统计学意义。
3.3 各组大鼠一氧化氮含量的变化 结果如图3所示:与生理盐水组和EPO 治疗组相比,AD 模型组的一氧化氮含量明显地升高(P <0.05),差异有统计学意义。

图1 各组大鼠总抗氧化能力的变化与生理盐水组和EPO治疗组相比,AD模型组T-AOC明显下降(P<0.05)
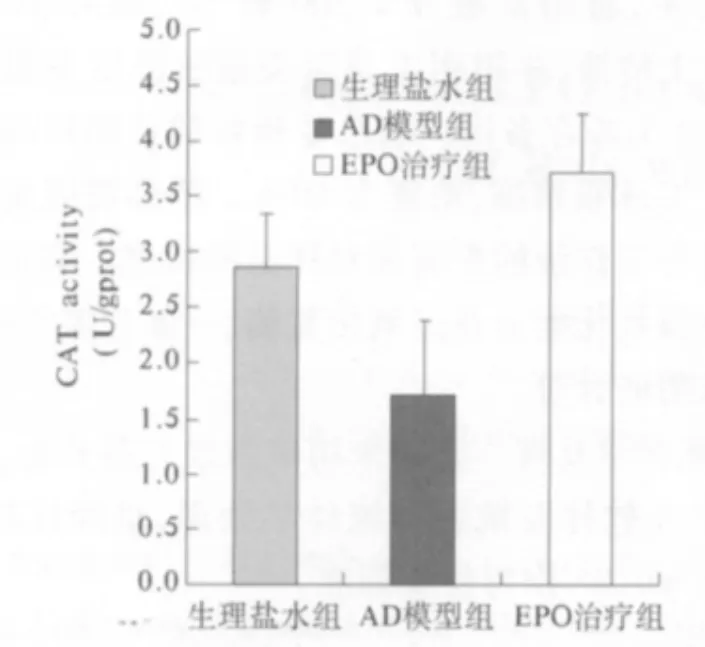
图2 各组大鼠过氧化氢酶活性的变化与生理盐水组和EPO治疗组相比,AD模型组的CAT活性明显下降(P<0.05)

图3 各组大鼠NO含量的变化与生理盐水组和EPO治疗组相比,AD模型组的NO含量明显地升高(P<0.05)
3.4 各组大鼠一氧化氮合成酶活性的变化 结果如图4 所示:与生理盐水组相比,AD 模型组的一氧化氮合成酶活性明显地升高(P <0.05),同时,EPO治疗组与AD 模型组相比,一氧化氮合成酶的活性显著地下降(P <0.05)差异具有统计学意义。
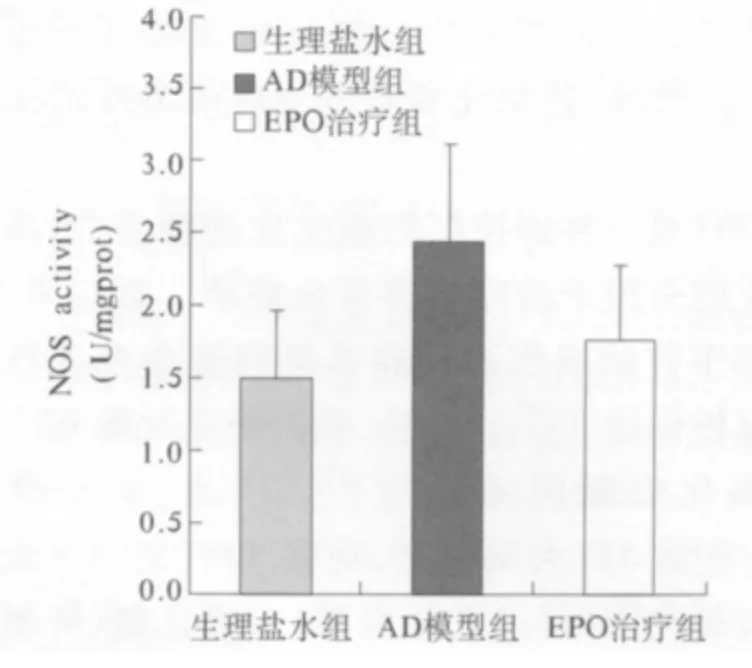
图4 各组大鼠NOS 活性的变化与生理盐水组和EPO 治疗组相比,AD 模型组的NOS 活性明显地升高(P <0.05)
4 讨论
许多研究认为氧化应激反应在阿尔茨海默病的发病过程中扮演了重要的角色[13-15]。首先,氧化应激反应可以诱导Aβ 的产生和聚集[16-17],氧自由基的产生可以引起Aβ 前体蛋白表达的增高[18],并增加神经细胞和神经胶质细胞增生内Aβ 的分泌[19]。此外,氧化应激反应还能增高早老素1[20]和Aβ 分泌酶[21]的表达。另一方面,Aβ 沉积也能够引起氧化应激反应和损伤[21-22],Aβ 通过过渡金属离子(如Cu2+,Zn2+,Fe3+)来刺激活性氧自由基的产生[23]。Aβ 沉积可以引起神经元细胞膜脂质的过氧化,导致膜功能受损,引发Ca2+内流,造成细胞内Ca2+超载,损伤细胞器,从而引起细胞毒性反应和神经元细胞的凋亡[24]。同时,Aβ 沉积所诱导的氧化应激反应还能够线粒体DNA 的突变,引起氧化呼吸链功能受损,使线粒体功能的紊乱,阻碍细胞的能量代谢通路,从而产生更多的活性氧自由基,造成细胞的损伤[25]。另外,Aβ 的聚集可以损伤机体内多种抗氧化酶的功能,引起机体抗氧化能力下降,加重氧化应激反应造成的损伤。
该实验通过大鼠海马区注射Aβ1-42制作AD 动物模型,并应用EPO 进行治疗,通过检测海马组织的总抗氧化能力(T—AOC)和过氧化氢酶(CAT)活性,来观察EPO 对AD 样大鼠海马组织抗氧化能力的影响。结果显示,AD 模型组与生理盐水组相比,T—AOC 和CAT 活性明显降低,说明Aβ 削弱了AD模型组大鼠多种抗氧化酶的活力,造成了AD 模型组大鼠抗氧化能力的下降。而EPO 治疗组与AD模型组相比,T—AOC 和CAT 活性显著地升高而NO 含量和NOS 活性下降,说明EPO 增强了机体内多种抗氧化酶的活力,从而有效地提高了机体总体的抗氧化能力,减轻了氧化应激反应对细胞的伤害,保护了神经元细胞。
[1]Selkoe D J. Alzheimer’s disease:Genes,proteins,and therapy[J]. PHYSIOLOGICAL REVIEWS,2001,81(2).
[2]Barkats M,Abrioux P,Mallet J. Overexpression of glutathione peroxidase increases the resistance of neuronal cells to Abeta-mediated neurotoxicity.[J]. Journal of neurochemistry,2000,75(4):1 438 -1 446.
[3]Boldogh I,Kruzel M L. Colostrinin (TM):An oxidative stress modulator for prevention and treatment of age - related disorders[J]. Journal of alzheimers disease,2008,13(3).
[4]Moon J H,Kim S Y,Lee H G,et al. Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar beta amyloid peptide (1 -42)-stimulated microglia[J]. Experimental and molecular medigine,2008,40(1):11-18.
[5]Sultana R,Mohmmad - Abdul H,Butterfield D A. Protective effect of the xanthate,D609,on Alzheimer’s amyloid beta-peptide (1 -42)-induced oxidative stress in primary neuronal cells[J]. Free radical research,2004,38(5):449 -458.
[6]Fisher J W. Erythropoietin:Physiology and pharmacology update[J]. Experimental biology and medicine,2003,228(1).
[7]Gonzalez F F,Mu D,Wendland M,et al. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke[J]. Developmental neuroscience,2007,29(4 - 5):321-330.
[8]Grasso G,Sfacteria A,Meli F,et al. Neuroprotection by erythropoietin administration after experimental traumatic brain injury[J]. Brain research,2007:99 -105.
[9]Lykissas M G,Vekris M D,Sakellariou E. The role of erythropoietin in central and peripheral nerve injury[J]. Clinical neurology and neurosurgery,2007,109(8):639 -644.
[10]Wu Y,Sun S,Yang W. Protective effect of erythropoietin against 1-methyl-4-phenylpyridinium-induced neurodegenaration in PC12 cells.[J]. Neuroscience bulletin,2007,23(3):156 -164.
[11]Cetin H,Oktem F,Uz E,et al. Novel evidence suggesting an anti-oxidant property for erythropoietin on vancomycin - induced nephrotoxicity in a rat model[J]. Clinical and experimental pharmacology and physiology,2007,34(11):1 181 -1 185.
[12]Katavetin P,Eiam-Ong S. Antioxidative effects of erythropoietin.[J]. 2007(107).
[13]Markesbery W R. Oxidative stress hypothesis in Alzheimer's disease.[J]. Free radical biology & medicine,1997,23(1):134-147.
[14]Pratico D. Oxidative injury in diseases of the central nervous system:focus on Alzheimer's disease.[J]. The American journal of medicine,2000,109(7):577 -585.
[15]Behl C. Alzheimer’s disease and oxidative stress:implications for novel therapeutic approaches. [J]. Progress in neurobiology,1999,57(3):301 -323.
[16]Tabner B J,Turnbull S,Paleologou K E,et al. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia[J]. Journal of biological chemistry,2005,280(43):35 789 -35 792.
[17]Murakami K,Ohigashi H,Nagao M,et al. Formation and stabilization model of the 42 -mer A beta radical:Implications for the long-lasting oxidative stress in Alzheimer's disease[J]. Journal of the american chemical society,2005,127(43):15 168 -15 174.
[18]Patil S,Masserang A. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons[J]. Neuroscience letters,2006,406(1 -2):55 -59.
[19]Murray I,Komatsu H,Xiao G,et al. Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins[J]. Journal of biological chemistry,2007,282(13):9 335 -9 345.
[20]Tamagno E,Guglielmotto M,Aragno M,et al. Oxidative stress activates a positive feedback between the gamma-and beta-secretase cleavages of the beta-amyloid precursor protein[J]. Journal of neurochemistry,2008,104(3):683 -695.
[21]Tong Y,Fung V,Qing H,et al. Oxidative stress potentiates BACE1 gene expression and A beta generation[J]. Journal of neural transmission,2005,112(3):455 -469.
[22]Tamagno E,Obbili A,Borghi R,et al. Oxidative stress increases expression and activity of BACE in NT2 neurons[J]. Neurobiology of disease,2002,10(3):279 -288.
[23]Huang X,Atwood C S,Tyndall J D,et al. Cu(II)potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction.[J]. The Journal of biological chemistry,1999,274(52):37 111 -37 116.
[24]Bezprozvanny I,Mattson M P. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease[J]. Trends in neurosciences,2008,31(9):454 -463.
[25]Wang J,Xie C,Lovell M A. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease[J]. Journal of neurochemistry,2005,93(4):953 -962.
猜你喜欢
山东冶金(2022年3期)2022-07-19
疯狂英语·初中天地(2022年5期)2022-07-06
疯狂英语·初中版(2022年5期)2022-05-11
中学生数理化·八年级物理人教版(2018年5期)2018-06-21
中华胃食管反流病电子杂志(2016年4期)2016-11-07
武大国际法评论(2016年2期)2016-06-01
中国造纸(2015年7期)2015-12-16
中国医学科学院学报(2015年5期)2015-03-01
中国当代医药(2015年17期)2015-03-01
现代检验医学杂志(2015年2期)2015-02-06
