
原发性色素沉着性结节性肾上腺皮质病2例报道并文献复习
2013-11-15 02:56李乐乐周晓涛谷伟军杨国庆吕朝晖巴建明母义明陆菊明李江源潘长玉
山西医科大学学报 2013年3期
李乐乐,周晓涛,谷伟军,杨国庆,杜 锦,吕朝晖,巴建明,母义明,陆菊明,李江源,潘长玉,
原发性色素性结节性肾上腺皮质病(primary pigmented nodular adrenocortical disease,PPNAD)是非ACTH依赖性库欣综合征的一种罕见亚型,其在库欣综合征中所占比例不超过1%[1],此病以双侧肾上腺皮质多发性自主分泌的色素沉着结节和结节间皮质组织萎缩为特征。由Chute等[2]于1949年首次描述,但直到20世纪80年代中期临床上才认识到它是库欣综合征的一种特殊表现类型。我院于1996年在国内首次报道了此病[3],迄今国内外报道例数仍较少,国内报道不足50例,临床上较为罕见,加之临床医师对其认识不足,多有误诊漏诊,以致延误诊治。现将我科收治的2例报道如下并结合现今国内报道的23例[4-9]患者的临床资料进行分析,复习相关文献,以提高临床医师对此病的认识。
1 资料和方法
详细介绍解放军总医院内分泌科收治的2例PPNAD患者的临床病例,并检索国内相关PPNAD病例报道文献(共检索出国内报道23例患者),从性别比例、发病年龄、病程、临床表现、实验室检查、影像学表现、病理特点、治疗及术后随访等方面进行回顾分析。
1.1 我院收治的2例患者的临床资料
病例1,男性,18岁。主因逐渐出现向心性肥胖、毳毛增多、痤疮、皮肤瘀斑和紫纹,伴有头晕、头痛、乏力和记忆力减退入院。入院后查血尿离皮质醇水平明显升高,大剂量地塞米松抑制试验F和UFC均未被抑制。肾上腺CT扫描示左侧肾上腺内支增粗,疑为腺瘤。考虑诊断为“库欣综合征,左侧肾上腺腺瘤”,并于1992-04在我院泌尿外科行左侧肾上腺切除术。术后病理报告为:左侧肾上腺体积明显增大,切面棕黄色,镜下可见多发、直径为0.3-2 mm的微结节,边界清楚,没有包膜。结节内细胞体积大,包浆丰富,嗜伊红染色,有脂质空泡,提示是类固醇合成功能活跃的束状带细胞,部分包浆内可见脂褐素沉着。术后症状一度缓解,半年后复发,1995-01再次入院。入院查血、尿皮质醇水平明显升高,大剂量地塞米松抑制试验未被抑制(见表1)。胸腰椎X线片显示普遍性骨质疏松。垂体增强CT和MRI扫描均未见垂体瘤。肾上腺CT扫描见右侧肾上腺增大,左侧缺如。经全科集体讨论,诊断为PPNAD。行右侧肾上腺全切术,术后病理支持PPNAD诊断。术后予氢化可的松50 mg/次,2次/d,口服,半年后复查症状逐渐好转,X线片骨质疏松较前明显改善。
病例2,女,18岁,汉族。2009年(15岁)起逐渐出现脸变圆、向心性肥胖、皮肤变薄,同时出现全身多处痤疮、毳毛增多,开始未引起重视,未予治疗。2011年因为腹痛就诊于当地医院诊断为“双侧输尿管结石”,同时测血清皮质醇34.50 μg/dl(正常值为5-28 μg/dl),双肾CT示双侧肾上腺稍增粗,左侧肾上腺外支似见结节样影,垂体MRI示未见异常,仍未予治疗。患者上述症状逐渐加重,2012-06-28入我院后查体血压152/102 mmHg、身高150 cm、体重 57 kg。化验检查示血ACTH、F节律紊乱,血皮质醇增高,血ACTH低,生化示血钾3.01 mmol/L,大小剂量地塞米松抑制试验均未被抑制。肾上腺CT示左侧肾上腺体积稍增粗,外侧支似可见5 mm结节影,增强扫描可见强化,右侧肾上腺未见明显异常(见图1)。双肾可见点状钙化影。垂体MRI未见明显异常。经全科讨论,考虑PPNAD可能性大,转泌尿外科于2012-07-23在全麻下行后腹腔镜下左肾上腺切除术,手术顺利。术后病理示:(左侧)肾上腺皮质多发结节状增生(图2,见封3)。切面灰黄灰褐色、质中,皮质带增厚。诊断明确为PPNAD。术后第6天转入我科复查血皮质醇仍旧高于正常,行大小剂量地塞米松抑制试验血尿皮质醇水平仍不受抑制(见表1),提示右侧肾上腺功能亢进,故需再次手术切除另一侧肾上腺。因考虑到左侧肾上腺全切术后不久,暂未予右侧肾上腺切除,嘱患者待身体状况恢复后再择期行右侧肾上腺全切。
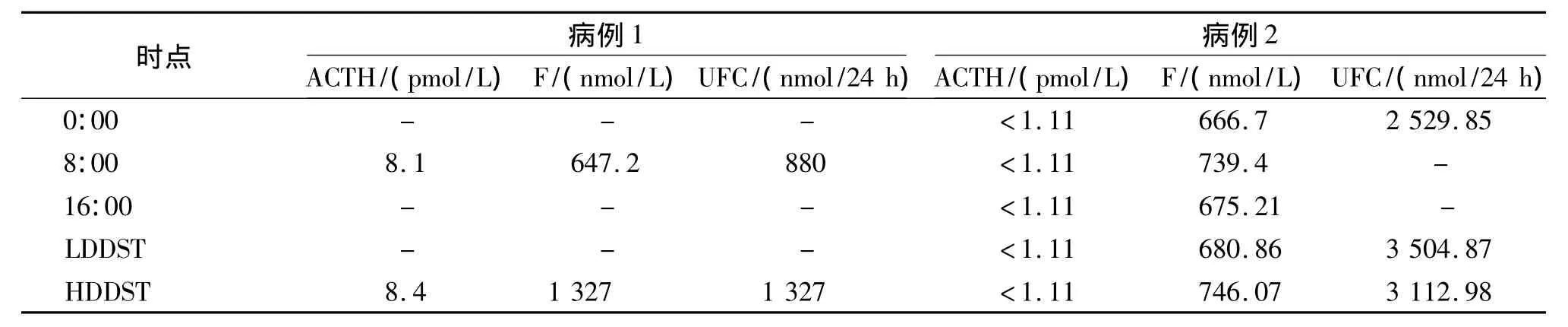
表1 我院2例患者术前皮质醇水平及大小剂量地塞米松抑制试验结果Tab 1 Results of high,low dose dexamethasone suppression tests and levels of adrenocorticotropic hormone before treatment in 2 cases of our hospital
1.2 国内25例患者临床资料总结
1.2.1 一般资料 25例患者性别比例、发病年龄、病程、典型临床表现、是否伴Carney综合征等资料见表2。25例患者确诊年龄11-53岁,年龄在40岁以上者仅1例。
1.2.2 实验室检查 25例患者中,1例因未就诊于内分泌科故未行地塞米松抑制试验,其余24例中仅1例患者皮质醇水平被抑制。协和医院报道15例患者中,有4例患者地塞米松抑制试验后皮质醇水平未下降反而升高,升高比例不超过50%。
1.2.3 影像学检查 25例患者均行肾上腺CT扫描,其中12例患者表现为结节样增粗,13例无异常改变。
1.2.4 病理学特点 23例患者均接受手术治疗,肾上腺病理可见色素沉积的结节样改变,支持PPNAD诊断。

图1 病例2患者肾上腺CT表现Fig 1 Adrenal CT features of case 2
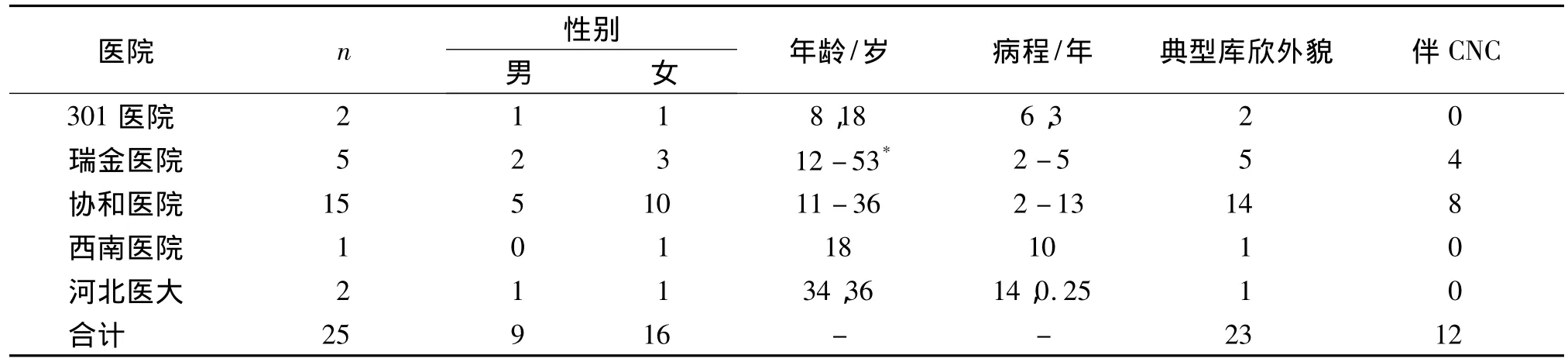
表2 国内报道23例患者临床资料Tab 2 Clinical data of 23 cases of PPNAD reported by China
2 讨论
2.1 PPNAD的病因及发病机制
PPNAD是非ACTH依赖性库欣综合征的一种罕见病因及表现形式。临床上可散发,也可作为Carney综合征的组成成分之一出现,这些患者约50%有家族聚集倾向,呈常染色体显性遗传。Sasano[10]等对其发病机制进行了阐述,他们认为 PPNAD患者双侧肾上腺结节来源于肾上腺皮质网状带,它们具有自主高分泌皮质醇激素功能,进而引发一系列临床表现。另有学者[11]通过光镜和电镜观察发现,PPNAD中不仅含丰富的嗜酸性细胞质的网状带细胞,还有大量富含脂质的空泡状束状带细胞,提示PPNAD结节可能来源于肾上腺皮质网状带和束状带。而在分子生物学方面,目前研究认为可能与位于17q22-24的cAMP依赖性蛋白激酶Aɑ调节基因(PRKAR1A)突变有关[12]。Horvath 等[13]对185个Carney综合征家系基因研究发现,约62%(114家系)存在有PRKAR1A基因突变,而在Carney综合征同时有PPNAD的患者中,该基因突变的检出率为80%。国内瑞金医院对1例Carney综合征进行家系调查及分子生物学研究,测序发现患者及其父亲均携带PRKAR1A基因突变[14]。近年来研究[15,16]发现除了 PRKAR1A 基因外,PPNAD 还与定位于2q31的磷酸二酯酶-11A(PDE11A)和定位于5q13的磷酸二酯酶-8B(PDE8B)基因突变相关。其确切发病机制有待于进一步研究。
2.2 临床特点
PPNAD虽为肾上腺自主分泌的库欣综合征,但症状相对较轻,部分患者呈亚临床状态,骨质疏松是其常见临床表现。除库欣综合征一般临床表现外,PPNAD尚具有以下特点:
①好发于青少年患者,高峰在20岁左右,4岁以内儿童和40岁以上成人病例罕见报道。我院2例患者发病年龄均在20岁以内,国内目前报道病例中发病最早者11岁[6],年龄在40岁以上者仅1例。女性多见,国内25例患者中男性9例,女性15例,男女比率为3∶5,与文献报道相符。
②大多数病例病程较长,呈隐匿性进展,从起病到确诊大约需要2-5年,可能与临床上对该病认识不足有关。我院第1例患者自发病到最终确诊历时6年,第一次曾误诊为“肾上腺腺瘤”。
③病情较轻,起病较隐匿,多数患者在确诊PPNAD之前曾被误诊。典型病例临床表现同皮质醇增多症,不典型病例可表现仅实验室检查提示有高皮质醇血症,非典型表现骨质疏松、肌无力等较为普遍。文献报道[17],在PPNAD患者中,约84%有典型库欣表现,10%缺乏典型表现,6%的患者仅实验室检查有异常。国内25例患者中23例有典型的库欣表现,占92%,略高于文献报道比例,可能与本研究所观察例数较少有关。我院所报道2例均有骨质疏松,考虑可能系病程较长,长期高皮质醇状态所致。
④最具特征性的表现是部分患者具有明显家族史,有学者报道,约90%的PPNAD与Carney综合征相关,PPNAD可作为Carney综合征的一项重要诊断标准。而在Carney综合征内分泌系统临床表现中,最多见的是 PPNAD,可发生于约25%的病例[18]。除PPNAD外,Carney综合征还往往伴面、颈部,躯干皮肤及口唇、结膜、巩膜着色斑及蓝痣,还可伴皮肤、乳房、心房黏液瘤,睾丸肿瘤,垂体生长激素瘤等。总结目前国内报道25例患者临床资料,得出伴Carney综合征者12例,所占比例为48%,提示我们在临床上很有必要对PPNAD患者常规筛查是否伴有Carney综合征,可通过检查患者有无皮肤黏膜色素沉着、心脏黏液瘤并调查家族史等协助诊断。
2.3 辅助检查
2.3.1 实验室检查 PPNAD患者血浆和尿游离皮质醇水平明显升高,皮质醇分泌失去昼夜节律性,血浆ACTH降低或测不出,大小剂量地塞米松抑制试验皮质醇不受抑制。国内25例患者中,1例因未于内分泌科就诊未行大小剂量地塞米松抑制试验,其余24例患者中,仅1例患者皮质醇水平被抑制。国外有学者[19]认为,口服地塞米松后UFC不被抑制反而升高,且升高幅度大于50%以上对PPNAD有诊断价值。他们通过对16例PPNAD患者和对照组肾上腺腺瘤患者行Liddle试验(实验操作类似国内大小剂量地塞米松抑制试验)后UFC反应水平后得出:24 h UFC较基础值升高50%以上对PPNAD诊断的特异性约为70%;而较基础值升高1倍则几乎可以确诊为PPNAD。该研究还得出这样一个结论:对临床上不典型、周期性库欣(PPNAD患者中较常见)患者,该实验对早期诊断有较高价值,可用于鉴别PPNAD。另有体外实验[20]提示:地塞米松可能通过作用于蛋白激酶A催化亚单位进而促进PPNAD肾上腺细胞糖皮质激素受体介导皮质醇激素释放增加。国内有关此方面的研究较少,仅协和医院[6]对此做过研究,在15例患者中仅4例抑制后皮质醇水平升高,但升高幅度小于50%,与文献报道不符,可能实验操作与国外不同,二者矛盾有待于我们进一步的临床研究。我院最近1例患者术前口服地塞米松后皮质醇水平不受抑制反而升高,但升高水平不超过基础值的50%。综上所述,就国内现状而言,该实验有效性仍待商榷。
2.3.2 影像学检查 对大多数患者而言,B超难以发现肾上腺病变。CT上一般无特异性改变,肾上腺可呈不规则增粗或小结节状,也可基本正常,临床上与肾上腺正常变异鉴别较为困难。据报道[17],有大约一半PPNAD患者影像学检查结果提示肾上腺形态无异常。况且18岁以上的PPNAD患者,部分在影像学上可表现为单侧2-3 cm的大结节样增生,其与腺瘤样增生的鉴别又成为临床工作中的一大挑战[17]。分析国内现有的25例患者的临床资料得出12例患者肾上腺CT表现为结节样增粗,13例无异常改变。因此,临床上单纯依据肾上腺影像学改变诊断PPNAD价值有限,但可协助除外肾上腺皮质腺瘤、ACTH非依赖性大结节样肾上腺增生(AIMAH)等病变。
2.3.3 病理学特点 PPNAD绝大多数为双侧肾上腺病变。双侧肾上腺可轻度增大,部分病例表现为正常大小,表面呈弥漫结节状改变,结节间界限清楚,结节直径1-3 mm,亦可见到直径为3 cm的较大结节。大体标本外观呈深褐色或有黑色素沉着,无包膜。结节间组织大多呈萎缩状态,细胞具有束状带细胞特征,而结节内细胞则具有网状带细胞的特点,包浆内富含脂褐素[5,6]。
2.4 诊断及鉴别诊断
PPNAD具有上述特征性组织形态改变,病理诊断容易,但因临床罕见,且影像学表现缺乏特异性,故而术前诊断较难。临床上应首先明确有无库欣综合征,再确定是否为肾上腺皮质自主分泌,通常依据典型实验室生化检查。对非ACTH依赖性库欣综合征,尤其是青少年且肾上腺CT无明显异常改变的患者,在除外外源性库欣综合征之后,PPNAD应被列入鉴别诊断,同时要考虑到伴发Carney综合征的可能。除与引起非ACTH依赖性库欣综合征的肾上腺皮质腺瘤、肾上腺皮质癌鉴别外,还应与以下疾病进行鉴别诊断:
2.4.1 非 ACTH依赖性大结节样肾上腺增生(AIMAH) 此病也是库欣综合征的罕见病因,也是不依赖于ACTH的双侧肾上腺增生病变,但该病患者肾上腺CT可见双侧肾上腺体积多明显增大,可见单个或多个大结节,正常肾上腺组织被扭曲。在某些患者可见肾上腺弥漫性增大而无显著的结节。与PPNAD显著不同的是后者肾上腺体积小,结节为多发性小结节。肾上腺CT有助于此二者鉴别。
2.4.2 异位皮质醇瘤 此病临床上亦表现为非ACTH依赖性库欣综合征,影像学上可见肾上腺正常大小或呈萎缩状态,易与PPNAD混淆。二者鉴别有赖于找到异位肾上腺组织。肾上腺皮质异位目前认为是由于肾上腺髓质细胞在向肾上腺皮质区域迁移的过程中,肾上腺皮质的碎片可能被分裂开来,有些与尿生殖嵴关系较为紧密的碎片在性腺迁移的过程中发生了异位。肾上腺皮质可异位于睾丸、精索、阔韧带、肾脏、腔静脉后、腹腔区域,其中32%异位于腹腔,23%异位于阔韧带,7.5%异位于附睾,3.8%-9.3% 异位于精索[21,22]。
2.4.3 色素性肾上腺皮质腺瘤(又称为黑色腺瘤)
黑色腺瘤最初被认为是无功能瘤,大多都是在尸检中发现,但随后发现部分患者可表现出高分泌功能,其中以皮质醇分泌增多为著,尚有原发性醛固酮增多症的报道。其增生的结节内细胞胞质也含有丰富的脂褐素,实验室检查也可表现为地塞米松抑制试验后皮质醇反常性升高,故需与PPNAD鉴别[23]。二者不同之处在于黑色腺瘤一般为单侧性,单个结节,常发生于皮髓质交界处。临床上此二者术前鉴别较为困难。确诊依赖于术后病理检查。
2.5 治疗及随访
PPNAD绝大多数是双侧肾上腺病变,手术切除肾上腺是治疗本病的有效方法,且术后应用糖皮质激素替代可避免出现 Nelson综合征[24]。文献报道[25],单侧肾上腺切除治愈率仅为30%,肾上腺次全切除术为60%,而肾上腺全切约为90%。从我院2例患者中也可看出,单侧肾上腺切除术仅可暂时缓解症状,血皮质醇节律较难恢复,ACTH仍受抑制,因而又追加对侧肾上腺全切。因此,如术前诊断明确,应尽量行双侧肾上腺全切。术前应用小剂量米托坦可安全有效地改善患者的高皮质醇状态[26]。
术后应每半年到1年随访一次,目的在于监测患者术后皮质醇水平变化、激素替代是否合理及发现潜在的其他Carney综合征病变。
综上,对影像学检查无明显异常改变的非ACTH依赖性库欣综合征患者,尤其是青少年患者,要常规将PPNAD列入鉴别诊断,同时要警惕伴发Carney综合征可能性。双侧肾上腺切除后予激素替代治疗是本病的最佳治疗方法,术后应密切随访。
[1] Berherat J.Carney complex(CNC)[J].Orphanet J Rare Dis,2006,1:21.
[2] 陈家伦.临床内分泌学[M].上海:上海科学技术出版社,2011:544-545.
[3] 李江源,高江平,母义明,等.原发性色素结节性肾上腺皮质异常增生型库欣综合征[J].解放军医学杂志,1996,21(5):395-396.
[4] 祝宇,吴瑜璇,周文龙,等.原发性色素性结节状肾上腺皮质病五例临床分析[J].中华外科杂志,2005,43(14):944-947.
[5] 陈中,段光杰,李苏,等.双侧原发性色素性结节状肾上腺皮质病1例报道及文献复习[J].临床与实践病理学杂志,2011,27(10):1131-1133.
[6] 张学斌,李汉忠,黄厚峰,等.原发性色素沉着性结节性肾上腺皮质病的临床诊治[J].临床泌尿外科杂志,2010,25(5):371-374.
[7] 周亚茹,李乃喆,杨建柱,等.原发性色素结节性肾上腺皮质病2例报告[J].中国实用内科杂志,2008,28(11):985-987.
[8] 蔚青,金晓龙,朱延波,等.原发性色素性结节状肾上腺皮质并的临床病理特征:附5例报道[J].诊断学理论与实践,2006,5(6):523-525.
[9] 李伟,冯凯,王鸥,等.8例原发性色素性结节状肾上腺皮质病的临床分析[J].基础医学与临床,2010,30(5):538-541.
[10] Sasano H,Miyazaki S,Sawai T,et al.Primary pigmented nodular adrenocortical disease(PPNAD):Immunohistochemical and in situ hybridisation analysis of steroidogenic enzymes in 8 cases[J].Mod Pathol,1992,5:23-29.
[11] Travis WD,Tsokos M,Doppman JL,et al.Primary pigmented nodular adrenocortical disease.A light and electron microscopic study of eight cases[J].Am J Surg Pathol,1989,13(11):921-930.
[12] Pereira AM,Hes FJ,Horvath A,et al.Association of the MIV PRKAR1A mutation with primary pigmented nodular adrenocortical disease in two large families[J].J Clin Endocrinol Metab,2010,95:338-342.
[13] Horvath A,Bertherat J,Groussin L,et al.Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A(PRKAR1A):an update[J].Hum Mutat,2010,31(4):369-379.
[14] 顾燕云,陈瑛,宋怀东,等.一例家族性Carney综合征临床及分子生物学研究[J].中华内科杂志,2004,43(10):764-768.
[15] Libe R,Horvath A,Vezzosi D,et al.Frequent phosphodiesterase 11A gene(PDE11A)defects in patients with Carney complex(CNC)caused by PRKAR1A mutations:PDE11A may contribute to adrenal and testicular tumors in CNC as a modifier of the phenotype[J].J Clin Endocrinol Metab,2011 ,96(1):E208-E214.
[16] Horvath A,Giatzakis C,Tsang K,et al.A cAMP-specific phosphodiesterase(PDE8B)that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues:a novel PDE8B isoform in human adrenal cortex[J].Eur J Hum Genet,2008,16(4):1245-1253.
[17] Lim LC,HC TL,Rajasoorya C.Unravelling the Mystery in a Case of Persistent ACTH-independent Cushing Syndrome[J].Ann Acad Med Singapore,2006,35(12):892-896.
[18] Manipadam MT,Abraham R,Sen S,et al.Primary pigmented nodular adrenocortical disease[J].J Indian Assoc Pediatr Surg,2011,16(4):160-162.
[19] Strataki C A,Sarlis N,Kirschner L S,et al.Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease[J].Ann Intern Med,1999,131(8):585-591.
[20] Louiset E,Strataki C A,Perraudin V,et al.The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits[J].J Clin Endocrinol Metab,2009,94(7):2406-2413.
[21] Ayala AR,Basaria S,Udelsman R,et al.Corticotropin-Independent Cushing syndrom caused by an ectopic adenoma[J].J Clin Endocrinol Metab,2000,85(8):2903-2906.
[22] Wang XL,Dou JT,Gao JP,et al.Laparoscope resection of ectopic corticosteroid-secreting adrenal adenoma[J].Neurol Endocrinol Lett,2012;33(3):265-267.
[23] Kamalanathan S,Mahesh DM,Muruganandham K,et al.Black adrenal adenoma:distinction from PPNAD[J].BMJ Case Rep ,2012:Epub.
[24] Powell AC,Stratakis CA,Patronas NJ,et al.Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia[J].Surgery,2008,143(6):750-758.
[25] 祝宇,吴瑜璇,芮文斌,等.原发性色素性结节状肾上腺皮质病(附4 例报告)[J].临床泌尿外科杂志,2003,18(5):271-273.
[26] Cignarelli M,Picca G,Campo M,et al.A six month mitotane course induced sustained correction of hypercortisolism in a young woman with PPNAD and Carney complex[J].J Endocrinol Invest,2005,28(1):54-60.
猜你喜欢
中国急救医学(2022年2期)2022-11-15
成都医学院学报(2021年2期)2021-07-19
中国民间疗法(2021年6期)2021-06-09
世界科学技术-中医药现代化(2021年10期)2021-03-02
小资CHIC!ELEGANCE(2021年46期)2021-01-11
睿士(2020年11期)2020-11-16
疯狂英语·新读写(2020年3期)2020-06-06
中国体育教练员(2017年2期)2017-07-31
海军医学杂志(2015年1期)2015-06-23
安徽医药(2014年5期)2014-07-07
