
利用伪氨基酸组分和支持向量机预测抗冻蛋白
2013-11-14 07:10:32许嘉
生物信息学 2013年4期
许 嘉
(内蒙古科技大学分析测试中心,内蒙古包头014010)
抗冻蛋白(Antifreeze protein,AFP)是一类能够特异性结合冰晶、提高生物抗冻能力的蛋白质[1]。这类蛋白最初是在南北极的海洋鱼类血清中发现,近年来,在昆虫、真菌、细菌和某些植物体内也均发现存在抗冻蛋白。这类蛋白通过与冰晶的特异性相互作用,阻止生物体内冰核的形成与生长,维持生物体内的溶液状态。因此,对抗冻蛋白的理论研究有助于揭示抗冻蛋白的活性和抗冻机理。
正确判断一条新测序的蛋白质是否为抗冻蛋白对于生物工程发展、作物的改造十分重要。然而,利用实验手段来判断是否是抗冻蛋白不但费时,而且会消耗很多资源。随着大量生物基因组测序的完成,海量基因组、蛋白质组、转录组数据的产生,利用机器学习算法来预测蛋白质的类型和功能不仅节约了实验成本,而且能够大大提高实验效率。后基因组时代为我们提供了大量蛋白质序列和注释信息,同时为理论预测抗冻蛋白提供了可能性[2]。
目前,已有一些判别方法用于抗冻蛋白的预测[3-4],且取得了一定的结果。然而,仍缺乏对抗冻蛋白有效的描述。本文利用伪氨基酸组分来描述抗冻蛋白序列,并利用支持向量机来对抗冻蛋白进行预测。
1 数据库
抗冻蛋白原始数据从 http://www3.ntu.edu.sg/home/EPNSugan/index_files/AFP-Pred.htm[3]下载。该数据集包含了481条抗冻蛋白序列和9 193条非抗冻蛋白序列,这些数据的序列一致性低于40%。如果正负数据集的数目偏差过大,会导致错误的评估预测模型。因此,为了平衡正负集数据,分别选取400条抗冻蛋白和400条非抗冻蛋白作为基准数据集,并进一步将正负数据集随机分为训练集和测试。这两集合分别包含200条抗冻蛋白和200条非抗冻蛋白。
2 预测算法
2.1 特征提取
伪氨基酸组分(PseAAC)[5]是 Chou教授提出的一种能够很好地表征蛋白质序列的信息参数。它不但能够描述蛋白质序列的氨基酸组成,而且能够描述蛋白质氨基酸序列的物理化学性质的关联。下面对伪氨基酸组分进行描述。
如果将一个氨基酸残基数为L的蛋白质X表示成,R1R2R3…RL那么,这条蛋白质序列就可以表示成由20+λ个离散数值定义的一个20+λ维向量,定义形式如下:
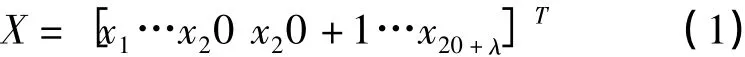
这里

其中,fi表示20种不同氨基酸残基在蛋白质X中出现的频率。ω是蛋白质序列关联的权重因子。通常,权重因子的选择范围定在ω=0.05到0.7之间,这里我们选取ω=0.05。θj是j阶序列相关系数:

公式(3)中相关性函数Θ(Ri,Ri+j)是可以由以下公式得出:
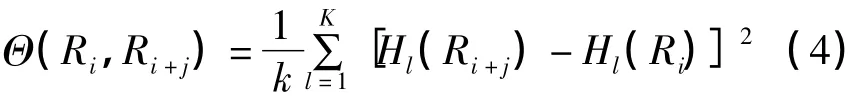
其中,k是因子个数,Hl(Ri)是第i个氨基酸残基所具有的任一种物理化学特征。这些物理化学特征主要包括亲水性,疏水性,侧链聚集度,a-COOH基的PK值,α-NH3+基的PK值,温度为25℃时的pI值。这些物化性质的值需经过标准化处理,公式如下:

这里Hl0(i)是第i个氨基酸残基物理化学特征值的原始值,可从网站 http://chou.med.harvard.edu/bioinf/PseAAC/获得。
2.2 支持向量机
支持向量机是一种优秀的机器学习方法,并已广泛运用于生物信息学的领域,比如:转录起始点和蛋白质亚细胞定位等多个方面。其优点在于能够同时最小化经验误差与最大化几何边缘区,因此支持向量机也被称为最大边缘区分类器。其基本思想是将向量映射到一个更高维的空间里,使得不同类型的向量在高维空间中线性可分。对于待分类样本,其判别函数具有如下形式:
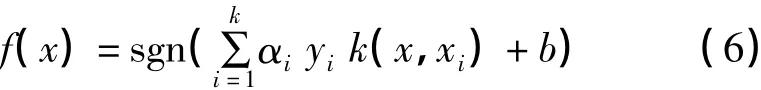
其中,k(x,xi)称为核函数,通过选取不同的核函数可以得到不同的支持向量机,常用的核函数有以下几种形式:
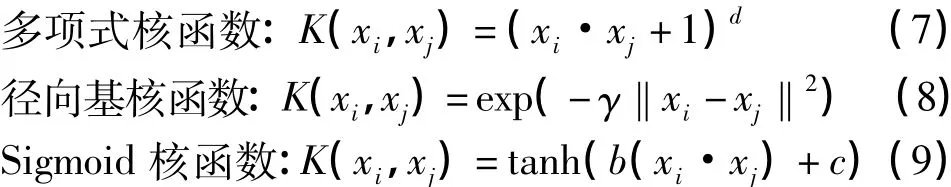
(6)~(8)式中,d、γ、b和 c分别为三种核函数的可调参数。本文采用由Chang和Lin开发的LIBSVM软件包[6],选取径向基函数(RBF)作为支持向量机的核函数,调整误差惩罚参数C及核函数参数γ,可得到最佳预测模型。这里使用LIBSVM中的gridsearch程序来优化参数C和γ。
2.3 精度估计
利用敏感性(Sensitivity,Sn)、特异性(Specificity,Sp)和总体准确率(Overall accuracy,OA)为评价指标测试模型的预测性能,其定义如下:
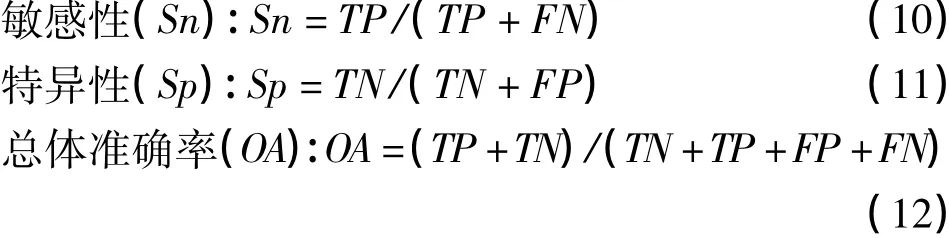
其中,TP、TN、FP和FN分别为正确预测抗冻蛋白数目,正确预测的非抗冻蛋白,非抗冻蛋白预测成为抗冻蛋白的数目和抗冻蛋白预测成非抗冻蛋白的数目。
3 结果与讨论
以伪氨基酸组分为特征,利用支持向量机进行分类。利用grid方法对训练集进行参数寻优,建立最优模型。发现当C=32 768且γ=0.001 953 125时,模型的预测精度最高,对训练集预测精度达到91.3%。为检验模型的推广能力,我们利用构建好的模型对400条测试序列进行预测,结果表明有78.8%的蛋白质被预测成功,其中75.1%的抗冻蛋白和83.6%的非抗冻蛋白能够被正确预测。该结果证明伪氨基酸组分可用于抗冻蛋白的预测。
AFP-Pred是第一款用于抗冻蛋白预测的软件[3],其构建基于300条抗冻蛋白和300条抗冻蛋白。通过使用随机森林算法对抗冻蛋白进行预测,对训练集的预测精度达到81.3%,对测试集的预测精度达到 83.4%。最近,Zhao Xiaowei等开发了AFP_PSSM来预测抗冻蛋白[4],对训练集的预测精度为82.7%,对测试集的预测精度达到93.0%。
尽管已有对测试集的预测精度高于本研究结果,但对于训练集,本研究结果仍具备优势。此外,这些方法大多使用了蛋白质序列的进化信息和预测的二级结构信息,这些信息的获得和提取比本研究使用的伪氨基酸组分要更加复杂。特别是当查询的数据库中没有待查询序列的同源序列时,进化信息将不可用;当二级结构预测软件错误的预测了蛋白质结构时,那么提取的二级结构信息也不可信。因此,只从蛋白质一级序列出发来预测抗冻蛋白,能够避免以上问题的出现。
尽管目前的研究结果还不十分令人满意,但随着蛋白质序列数据库的不断充实,将考虑更多的信息,如寡肽频率、氨基酸约化等信息,以期提高分类模型的预测准确率。
References)
[1] Carvajal-Rondanelli PA,Marshall SH,Guzman F.Antifreeze glycoprotein agents:structural requirements for activity[J].Journal Science Food Agricuture,2011,91(14):2507-2510.
[2] Garner J,Harding MM.Design and synthesis of antifreeze glycoproteins and mimics[J].Chembiochem,2010,11(18):2489-2498.
[3] Kandaswamy KK,Chou KC,Martinetz T,Möller S,Suganthan PN,Sridharan S,Pugalenthi G.AFP-Pred:A random forest approach for predicting antifreeze proteins from sequence-derived properties[J].Journal of Theoretical Biology,2011,270(1):56-62.
[4] Zhao Xiaowei,Ma Zhiqiang,Yin Minghao.Using support vector machine and evolutionary profiles to predict antifreeze protein sequences[J].Interntional Journal of Molecular Science,2012,13(2):2196-2207.
[5] Chou KC.Prediction of protein cellular attributes using pseudo-amino acid composition[J].Proteins,2001,43(3):246-255.
[6] Fan RE,Chen PH,Lin CJ.Working set selection using the second order information for training SVM[J].Journal of Multivariate Analysis,2005,6:1889-1918.
猜你喜欢
新高考·高一数学(2022年3期)2022-04-28 07:02:46
煤气与热力(2021年12期)2022-01-19 05:19:30
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:24
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19 08:28:36
中国洗涤用品工业(2019年4期)2019-05-11 09:27:18
中成药(2018年8期)2018-08-29 01:28:26
中成药(2018年2期)2018-05-09 07:20:09
中成药(2018年1期)2018-02-02 07:20:05
高中生学习·高三版(2016年9期)2016-05-14 09:12:05
新高考·高二数学(2015年11期)2015-12-23 18:17:44
