
顶空气相色谱法测定空心胶囊中环氧乙烷残留量*
2013-10-25 01:55:44赵振午孙瑞贞胡永钢张生万
浙江师范大学学报(自然科学版) 2013年2期
赵振午, 孟 颖, 孙瑞贞, 马 丽, 胡永钢, 张生万
(1.山西大学 生命科学学院,山西 太原 030006;2.山西大学 化学化工学院,山西 太原 030006)
顶空气相色谱法测定空心胶囊中环氧乙烷残留量*
赵振午1, 孟 颖1, 孙瑞贞2, 马 丽1, 胡永钢2, 张生万1
(1.山西大学 生命科学学院,山西 太原 030006;2.山西大学 化学化工学院,山西 太原 030006)
针对2010年版中国药典规定的顶空气相色谱法测定空心胶囊中环氧乙烷残留量的干扰物问题,使用气相色谱-质谱(GC-MS)联用技术进行了系统的考察,发现干扰物主要来自实验用水中溶解的CO2,对实验用水在使用前加热煮沸即可有效地去除干扰.同时,选用RTX-WAX色谱柱,在柱温40 ℃,载气流量0.8 mL/min时分离效果和峰型最好.在此基础上对药典方法进行了改进,实验结果表明:环氧乙烷含量为0.21~12.67 μg时呈良好的线性关系,相关系数r=0.999 9;最低检出限为0.03 μg,加标回收率为98.6%~103.2%,相对标准偏差为0.7%~1.7%.
空心胶囊;环氧乙烷;干扰物;气相色谱-质谱联用;顶空气相色谱
空心胶囊生产过程中大多采用环氧乙烷(EO)灭菌.环氧乙烷是一种气态广谱杀菌剂,可以杀灭各种微生物,包括细菌、繁殖体、芽孢、病毒等[1],因其具有效果好、持效期长、操作简单等特点,目前已广泛应用于医药领域[2-4].然而,过量的环氧乙烷对人体毒害较大,不仅会引起急性中毒,还具有致过敏、致突变和致癌作用[5-7].为此,2010版中国药典规定环氧乙烷在空心胶囊中的残留量应小于0.000 1%[8].
目前,文献报道的空心胶囊中环氧乙烷的测定方法主要有2类:一是张雅青等[9]利用玻璃填充柱气相色谱法测定空心胶囊中环氧乙烷残留量,该方法利用填充柱直接进样,分离效果较差,不能使杂质和环氧乙烷较好地分离;另一为2010版药典规定的顶空气相色谱法测定空心胶囊中环氧乙烷残留量的标准检测方法[8].笔者在使用药典方法测定空心胶囊中环氧乙烷残留量的过程中,发现即便样品无环氧乙烷存在时仍能检出一定量的环氧乙烷,造成空心胶囊中环氧乙烷残留量偏高.为此,使用气相色谱-质谱(GC-MS)联用技术对顶空气相色谱法测定空心胶囊中环氧乙烷残留量的干扰物进行了系统考察,发现了干扰物及其主要来源,并对色谱柱及色谱条件进行了选择和优化,在此基础上对药典方法进行了改进.该方法用于山西广生胶囊有限公司生产的空心胶囊中环氧乙烷残留量的检测,取得了满意的结果.
1 实验部分
1.1主要仪器与试剂
Agilent 7890-5975C型GC/MS联用仪(配有FID检测器),Agilent 7694顶空进样器(美国Agilent公司),色谱柱分别为RTX-WAX(30 m×0.25 mm×0.5 μm ),BP21(25 m×0.32 mm×0.25 μm)和RTX-1(30 m×0.25 mm×0.25 μm),20 mL顶空瓶.环氧乙烷(含量≥98%,上海天莲精细化工有限公司).空心胶囊(山西广生胶囊有限公司).
1.2实验方法
1.2.1 实验用水的制备
将蒸馏水加热煮沸30 min后,加盖冷却,储存备用.
1.2.2 样品的配制
称取空心胶囊2.00 g,剪碎,加入20 mL顶空瓶中,加10.00 mL实验用水,加盖密封,摇匀待测.
1.2.3 环氧乙烷溶液的配制
1)环氧乙烷贮备液:取30 mL实验用水于50 mL容量瓶中,准确称量;在0 ℃以下移取环氧乙烷约0.15 mL,加入容量瓶中,不加瓶塞振摇,盖好瓶塞称量;前后2次称量之差得环氧乙烷质量为0.070 4 g,用实验用水定容至刻度,得1.41 mg/mL环氧乙烷贮备液.
2)环氧乙烷标准溶液:准确移取0.75 mL环氧乙烷贮备液于50 mL容量瓶中,用实验用水定容至刻度;取此溶液5.00 mL再稀释至50 mL,得2.11 μg/mL环氧乙烷标准溶液.
1.3分析方法
1.3.1 气相色谱分析条件
RTX-WAX(30 m×0.25 mm×0.5 μm)色谱柱;柱温40 ℃,进样口温度250 ℃,检测器温度250 ℃;载气He,流量0.8 mL/min;分流比10∶1;顶空平衡温度80 ℃,平衡时间15 min,定量环温度90 ℃,传输线温度100 ℃;定量环体积1 mL.
1.3.2 气/质联用分析条件
色谱分析条件同上.
质谱分析条件:电子轰击离子源(EI);电子能量70 eV;离子源温度250 ℃;传输线温度280 ℃;四级杆温度150 ℃;质量扫描范围为18~500;数据采集速率为1.56 scans/s;色谱柱为HP-5(30 m×0.25 mm×0.25 μm);载气He.
2 结果与讨论
2.1色谱条件选择与优化
2.1.1 色谱柱的选择
在色谱条件下,用药典规定范围内的RTX-WAX,BP21和RTX-1三种色谱柱分别对环氧乙烷标准液进行了测定[8],其结果见表1.从表1可知,使用色谱柱RTX-WAX的分离效果优于色谱柱RTX-1和BP21.所以选用RTX-WAX为实验用色谱柱.

表1 不同色谱柱所得环氧乙烷峰与邻近峰比较
2.1.2 载气流量的选择
在柱温45 ℃时,用RTX-WAX色谱柱分别对载气流量为0.6,0.8,1.0,1.5,2.0 mL/min进行了考察,结果见表2.表2数据表明,环氧乙烷峰与邻近峰在载气流量0.6 mL/min时分离效果最好,但其峰形相对较差.为保证定量分析的准确度,所以选择载气流量为0.8 mL/min.

表2 不同载气流量下环氧乙烷峰与邻近峰比较
2.1.3 柱温的选择
在已选定的色谱条件下,对柱温为35,40,45 ℃进行了考察,结果见表3.表3数据表明,环氧乙烷峰和邻近峰的分离效果在柱温35 ℃最好,但这时环氧乙烷的峰形比40 ℃时差.所以本实验选择柱温为40 ℃.选定条件下的色谱图见图1.

表3 不同柱温下环氧乙烷峰与邻近峰比较
2.2干扰物考察
2.2.1 干扰物来源
在上述选定的色谱条件下,对环氧乙烷标准溶液、未用环氧乙烷灭菌的空心胶囊及蒸馏水进行了测定,结果见图1.由图1可知,在未经环氧乙烷灭菌的空心胶囊(b)和蒸馏水(c)的色谱图中,与环氧乙烷保留时间(3.46 min)相同处仍有一峰存在,表明干扰物主要来源于实验用水(蒸
馏水),该干扰物对环氧乙烷残留量的测定会产生影响.

1:空气峰;2:杂质峰;3:环氧乙烷峰;4:干扰物峰a:环氧乙烷标准液;b:未用环氧乙烷灭菌空心胶囊;c:蒸馏水图1 环氧乙烷标准溶液等的色谱图
2.2.2 干扰物表征
按气/质谱联用分析条件,对蒸馏水及环氧乙烷溶液进行GC-MS分析,得到图2所示的总离子流色谱图和图3所示的质谱图.
由图2可知,在气/质联用分析条件下,蒸馏水的测定仍然得到与单独色谱分析基本相同的结论,即在环氧乙烷保留时间相同处仍有干扰物质存在.对图2的空气峰和蒸馏水中干扰物峰的质谱图(见图3)进行比较,结果表明,图3(a)和图3(b)中成分相同(m/z28为N2,m/z32为O2,m/z40为Ar,m/z44为CO2),所不同的是各离子峰相对强度不同,即各种气体的相对含量不同,尤其是图3(b)中m/z44的CO2相对含量有所增加,表明干扰峰可能是由于蒸馏水中溶解的CO2所致.
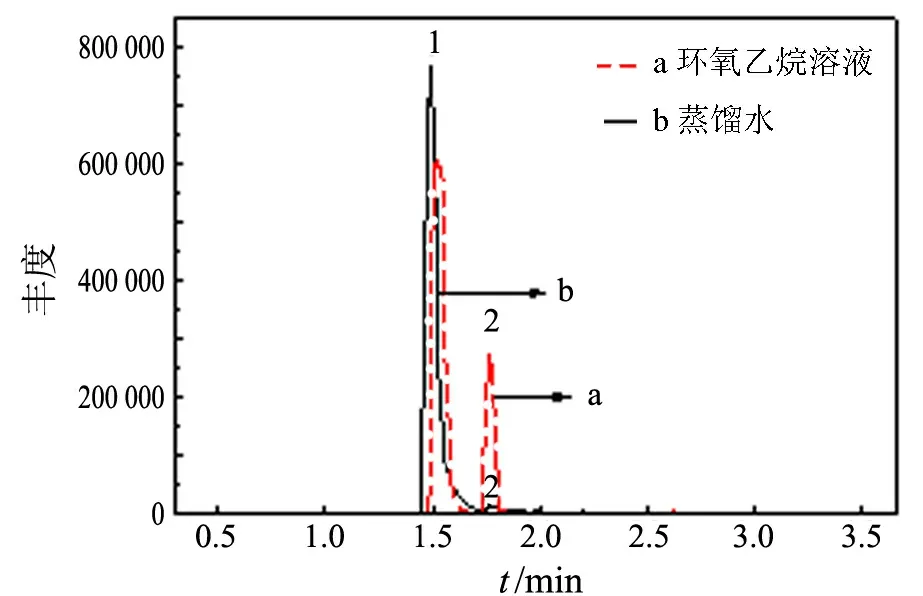
1:空气峰;2:环氧乙烷峰图2 环氧乙烷溶液及蒸馏水的总离子流色谱图

(a)空气峰 (b)蒸馏水中干扰物峰图3 空气峰和蒸馏水中干扰物峰的质谱图
2.2.3 干扰物消除
为确认上述GC-MS联用分析结果,在选定色谱条件下分别对蒸馏水、加热煮沸30 min的蒸馏水和用高锰酸钾重蒸过的蒸馏水进行了分析比较,结果如图4所示.
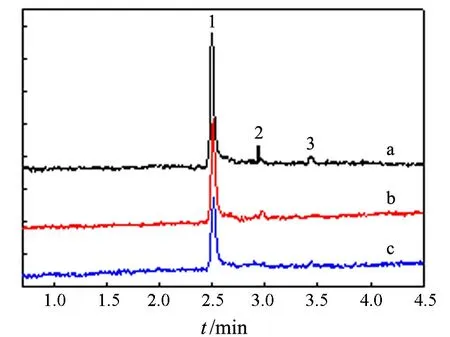
1:空气峰;2:杂质峰;3:干扰物峰a:蒸馏水;b:煮沸过的蒸馏水;c:高锰酸钾重蒸水图4 实验用水的色谱图
从图4可知,加热煮沸过的蒸馏水和高锰酸钾重蒸水,干扰峰(保留时间3.46 min)都可有效去除.这一结果进一步表明,干扰物的确来源于溶解在水中的CO2,通过加热煮沸蒸馏水即可消除干扰,并比高锰酸钾重蒸水易于制备.所以,实验用水选用加热煮沸过的蒸馏水.
2.3线性范围测定
准确移取2.11 μg/mL环氧乙烷标准溶液0.10,0.50,1.00,2.00,4.00和6.00 mL于6个20 mL顶空瓶中,再分别准确量取实验用水9.90,9.50,9.00,8.00,6.00和4.00 mL,配成待测样品溶液,其中环氧乙烷含量分别为0.21,1.06,2.11,4.22,8.45和12.67 μg,平行配置3份,在选定的色谱条件下进行测定.以峰面积为纵坐标,环氧乙烷含量为横坐标,绘制标准曲线,得回归方程Y=25 761X+2 108.5,r=0.999 9.结果表明,环氧乙烷含量为0.21~12.67 μg时呈良好的线性关系.以3倍噪声比计算,环氧乙烷的检出限为0.03 μg.
2.4精密度实验
在6个20 mL顶空瓶中,分别加入2.32 μg/mL环氧乙烷溶液1.00 mL,再分别准确加入9.00 mL实验用水,密封、摇匀,分别进行色谱测定,结果如表4所示.从表4可知,相对标准偏差为1.1%,表明本方法精密度良好.

表4 精密度测定结果
2.5重复性实验
取同一批号(211050419)空心胶囊6份,按1.2.2方法制备样品,对环氧乙烷残留量进行色谱测定,结果如表5所示.从表5数据可知,相对标准偏差为1.5%,表明本方法重复性良好.

表5 环氧乙烷重复测定结果
2.6加标回收率实验
对同一批号(211050419)空心胶囊,每份准确称取2.00 g,取15份,分别置于15个20 mL顶空瓶中,其中:1,2,3,4,5号瓶各准确加入2.99 μg/mL环氧乙烷溶液1.00 mL,再加入实验用水9.00 mL;6,7,8,9,10号瓶各准确加入4.20 μg/mL环氧乙烷溶液1.00 mL,再加入实验用水9.00 mL;11,12,13,14,15号瓶各准确加入5.75 μg/mL环氧乙烷溶液1.00 mL,再加入实验用水9.00 mL,密闭封口,摇匀,进行色谱测定,结果如表6所示.从表6可以看出,回收率为98.6%~103.2%,相对标准偏差为0.7%~1.7%,表示本方法满足检测需求.

表6 加标回收率测定结果
2.7样品分析
取批号210111759,211040149和211050419的3批空心胶囊产品,每批产品按1.2.2制样方法平行制备3份样品,分别用本方法(检测用水为煮沸过的蒸馏水)和药典方法(检测用水为未处理的蒸馏水)进行色谱测定,结果见表7.2010版中国药典2部明胶空心胶囊的环氧乙烷检测项[8规定:供试品溶液(2.00 g胶囊加10.00 mL水制成的顶空样品)中环氧乙烷的峰面积,不得大于对照品溶液(取2.00 μg/mL环氧乙烷溶液1.00 mL加9.00 mL水制成的顶空样品)中环氧乙烷的峰面积.从表7可知:药典方法比本方法的测定结果偏高;两种方法的结果都是批号211040149产品不合格,其余2批产品均符合药典要求.
3 结 论
采用GC-MS发现环氧乙烷残留量测定中的干扰物主要来自实验用水中溶解的CO2,对实验用水在使用前加热煮沸,即可有效去除干扰.同时,选用RTX-WAX色谱柱,在柱温40 ℃,载气流量0.8 mL/min的分离效果和峰形较好.在此基础上对药典方法进行了改进,该方法用于空心胶囊中环氧乙烷残留量的检测,取得了满意的结果.

表7 空心胶囊中环氧乙烷的测定结果
[1]杨君,王燕萍,王敏荣.顶空气相色谱法检测一次性医疗用品中环氧乙烷残留量[J].中国卫生检验杂志,2010,20(6):1345-1346.
[2]Thomas R Z,Ruben J L,ten Bosch J J,et al.Effect of ethylene oxide sterilization on enamel and dentin demineralization in vitro[J].J Dent,2007,35(7):547-551.
[3]Azzam M G,Roy M E,Whiteside L A.Second-generation locking mechanisms and ethylene oxide sterilization reduce tibial insert backside damage in total knee arthroplasty[J].J Arthroplasty,2011,26(4):523-530.
[4]Archer E,Allen H,Hopwood A,et al.Validation of a dual cycle ethylene oxide treatment technique to remove DNA from consumables used in forensic laboratories[J].Forensic Sci Int Genet,2010,4(4):239-243.
[5]张霞,周雯,王有森.环氧乙烷在灭菌物品中残留量测定及毒性研究进展[J].中国消毒学杂志,2005,22(2):217-218.
[6]Natarajan A T,Preston R J,Dellarco V,et al.Ethylene oxide:evaluation of genotoxicity data and an exploratory assessment of genetic risk[J].Mutat Res,1995,330(1/2):55-70.
[7]Melnick R L,Sills R C.Comparative carcinogenicity of 1,3-butadiene,isoprene,and chloroprene in rats and mice[J].Chem Biol Interact,2001,135/136:27-42.
[8]国家药典委员会.中华人民共和国药典:二部[M].2010年版.北京:中国医药科技出版社,2010:1204-1205.
[9]张雅青,李平.气相色谱法测定空心胶囊中环氧乙烷残留量[J].江西医药,1987,22(5):459-461.
(责任编辑 薛 荣)
DeterminationofresidualethyleneoxideinhollowcapsulesbytheheadspaceGC
ZHAO Zhenwu1, MENG Ying1, SUN Ruizhen2, MA Li1, HU Yonggang2, ZHANG Shengwan1
(1.SchoolofLifeScience,ShanxiUniversity,TaiyuanShanxi030006,China; 2.CollegeofChemistryandChemicalEngineering,ShanxiUniversity,TaiyuanShanxi030006,China)
Distractors in the determination of the residual ethylene oxide in hollow capsules by the headspace gas chromatography in the Chinese Pharmacopoeia of 2010 edition was systematically studied by GC-MS. Interference source was examined and found that interfering substances mainly came from CO2which dissolved in the experiment water and the boiled distilled water interference could be removed. Using RTX-WAX column in the column temperature of 40 ℃ and carrier gas flow 0.8 mL/min the ethylene oxide separated well and the peak pattern was preferably. The results showed that the concentration of ethylene oxide in the range of 0.21~12.67 μg had good linear relationship. The correlation coefficient was 0.999 9. The limit of detection was 0.03 μg/mL. The recoveries ranged from 98.6% to 103.2% and the relative standard deviation was from 0.7% to 1.7%.
hollow capsules; ethylene oxide; distractors; GC-MS; headspace GC
O656.31
A
1001-5051(2013)02-0193-05
2013-03-05
国家自然科学基金资助项目(31171748);山西省特色重点学科基金资助项目;山西省研究生优秀创新项目(20123015)
赵振午(1985-),男,山西忻州人,硕士研究生.研究方向:食品化学.
张生万.E-mail: zswan@sxu.edu.cn
猜你喜欢
广州化工(2020年20期)2020-11-02 03:02:52
Advances in Petroleum Exploration and Development(2019年1期)2019-08-05 09:23:32
石油炼制与化工(2016年5期)2016-04-06 21:46:49
分析化学(2015年6期)2015-06-18 10:28:17
化学反应工程与工艺(2015年1期)2015-04-16 03:06:11
机电信息(2014年35期)2014-02-27 15:54:29
机电信息(2014年26期)2014-02-27 15:53:34
首都食品与医药(2013年24期)2013-10-19 11:56:34
石油化工技术与经济(2013年6期)2013-04-09 02:00:51
中国石油石化(2010年19期)2010-11-07 08:16:56
