
加工方法对百合质量的影响研究△
2013-09-26 10:12:46聂慧严辉钱大玮段金廒欧阳臻钱叶飞管汉亮
中国现代中药 2013年4期
聂慧,严辉,钱大玮*,段金廒,欧阳臻,钱叶飞,管汉亮
(1.江苏大学 药学院,江苏 镇江 212013;2.南京中医药大学 江苏省方剂高技术研究重点实验室,江苏 南京 210046)
中药工业
加工方法对百合质量的影响研究△
聂慧1,2,严辉2,钱大玮2*,段金廒2,欧阳臻1,钱叶飞2,管汉亮2
(1.江苏大学 药学院,江苏 镇江 212013;2.南京中医药大学 江苏省方剂高技术研究重点实验室,江苏 南京 210046)
目的:研究加工方法对百合质量的影响,为百合加工方法的规范化提供参考依据。方法:新鲜百合样品分别采用煮制和蒸制2种烫片方法、不同干燥方法(包括热风烘干、微波干燥、远红外干燥和冷冻干燥)进行加工,样品中酚性甘油酯类、核苷、氨基酸及多糖等成分分别采用超高效液相-二极管阵列检测器(UPLC-DAD)、超高效液相-三重四级杆质谱联用仪(UPLC-TQ/MS)及紫外-可见分光光度法(UV)测定。结果:烫片过程,煮5~8 min和蒸8~10 min样品不仅外观色泽较佳,而且各成分含量较高;干燥方法,60~80 ℃烘干和冷冻干燥法较好。结论:烫片是百合干加工过程中必不可少的环节;确定最佳工艺条件为煮5~8 min,60~80 ℃烘干或冷冻干燥。
百合;烫片;干燥;工艺优化
百合味甘,性寒。具有养阴润肺,清心安神功能,用于阴虚燥咳、虚烦惊悸、失眠多梦、精神恍惚[1-2]。现代研究表明,百合含酚性甘油酯、多糖、甾体皂苷和生物碱等活性成分,具有抗疲劳、抗肿瘤、降血糖、抗氧化、免疫调节、镇静及抗应激损伤作用[3]。此外还含有氨基酸、核苷、微量元素、挥发油及磷脂等营养成分,常作为饮料及营养保健食品的基质原料应用于食品工业[4]。
百合多以百合干的形式贮藏与应用。传统百合干加工流程为:鲜百合选料→剥片→清洗→烫片→冷却→干燥→包装储藏。加工过程中烫片和干燥2个环节对其质量的影响较大,本研究以鲜百合为材料,研究烫片工艺与干燥过程中百合化学成分的变化,以优化各加工参数,为优质百合的生产提供科学依据。
1 仪器与试药
1.1 仪器
ACQUITY UPLC系统(二元高压泵,自动进样器,柱温箱,二极管阵列检测器,Waters公司);Xevo TQ质谱系统(Waters公司);MassLynxTM质谱工作站(Waters公司);UV-2000紫外-可见分光光度计(北京莱伯泰科仪器有限公司);Sartorius BT125D电子分析天平(德国塞利多斯公司);EPED超纯水系统(南京易普达易科技发展有限公司);KQ-250E型超声波清洗器(昆山禾创超声仪器有限公司);Anke GL-16GⅡ型离心机(上海安亭科学仪器厂)。
1.2 试药及试剂
1-O-阿魏酰甘油(Fer)、1-O-p-香豆酰甘油(Cou)、王百合苷D(Reg D)、王百合苷C(Reg C)、王百合苷A(Reg A)、王百合苷E(Reg E)均为本实验室从百合鳞叶中分离自制,其化学结构经1H-NMR、13C-NMR及ESI-MS分析确认,纯度经HPLC-UV检测均大于98%。
鸟嘌呤(Gua,批号:140631-200904)、黄嘌呤(Xan,批号:140662-200301)、胸腺嘧啶(Thym,批号:140708-200401)、腺嘌呤(Ade,批号:886-200001)、肌苷(Ino,批号:140669-200903)、环磷酸腺苷(Camp,批号:140709-200602)和葡萄糖(Glc,批号:0833-9501)购自中国食品药品检定研究院,供含量测定用。
胞苷(Cytd,批号:0001446223)、2′-脱氧胞苷(2′B,批号:1000851526)、2′-脱氧腺苷(2′X,批号:1000723530)、2′-脱氧鸟苷(2′Niao,批号:1000814345)、2′-脱氧尿苷(2′N,批号:1000943453)、胸苷(Thyd,批号:1000735425)、甘氨酸(Gly)、γ-氨基丁酸(GABA)、亮氨酸(Leu)、甲硫氨酸(Met)、苯丙氨酸(Phe)、色氨酸(Trp)、丙氨酸(Ala)、苏氨酸(Thr)、丝氨酸(Ser)、天冬酰胺(Asn)、谷氨酰胺(Gln)、谷氨酸(Glu)、瓜氨酸(Cit)、脯氨酸(Pro)、缬氨酸(Val)、酪氨酸(Tyr)、羟脯氨酸(Hpro)、异亮氨酸(Ile)、组氨酸(Hit)、精氨酸(Arg)购自Sigma公司,其纯度经HPLC检测均大于98%;乙腈(德国Merck公司)为色谱纯,其他试剂均为分析纯。
实验用百合样品于2012年10月采自安徽省霍山县,经南京中医药大学段金廒教授鉴定为百合科植物卷丹LiliumlancifoliumThunb.的新鲜肉质鳞叶。凭证标本存放于南京中医药大学江苏省方剂高技术研究重点实验室。
2 方法与结果
2.1 样品制备
新鲜百合样品,经选料、剥片、清洗后分别进行烫片工艺和干燥方法研究。样品信息见表1、2。
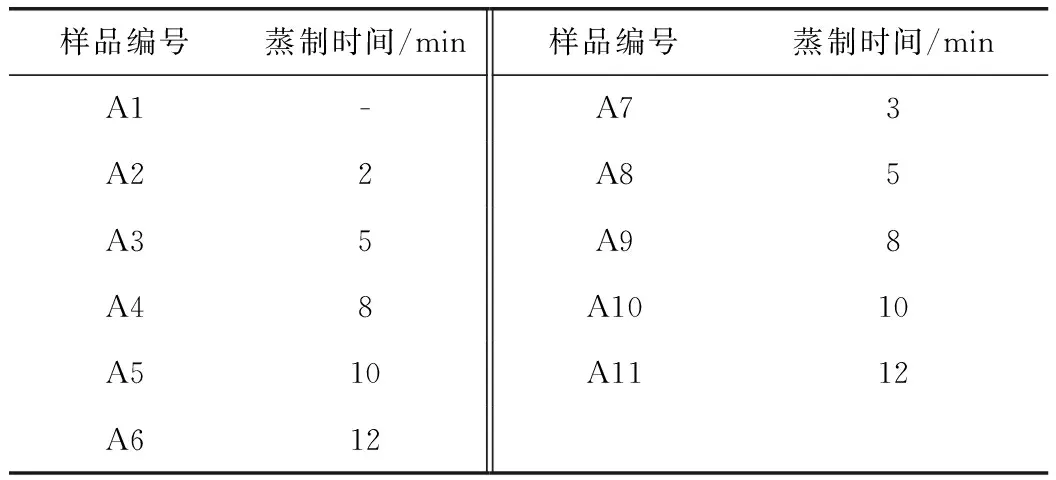
表1 烫片样品信息表
注:A1为鲜品
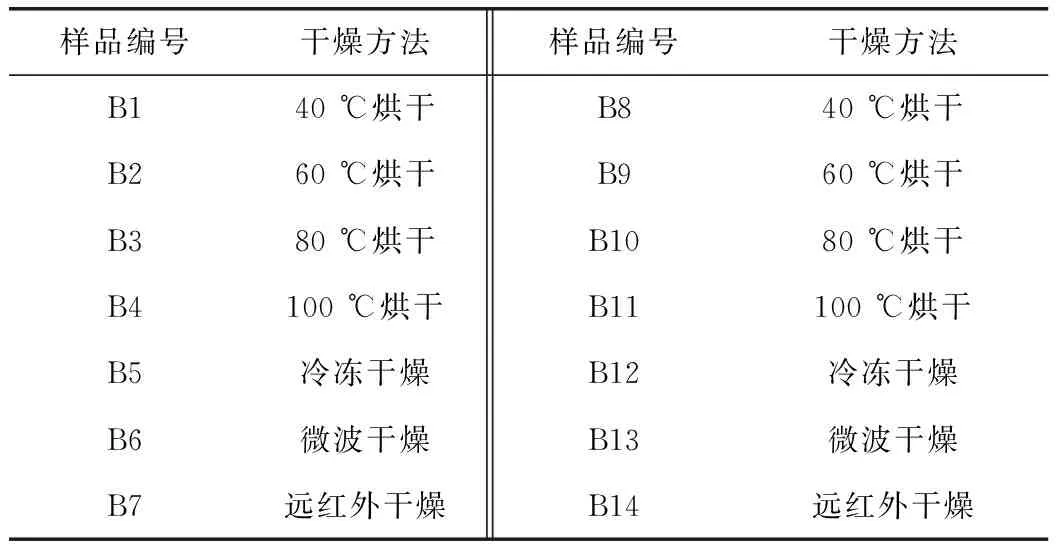
表2 干燥样品信息表
注:B1~B7为鲜品,B8~B14为煮制品
2.2 酚性甘油酯类成分测定[5-6]
2.2.1 供试品溶液的制备
2.2.1.1 烫片样品供试品溶液的制备 取样品约3 g,精密称定,加适量50%乙醇,匀浆,置于具塞锥形瓶,加50%乙醇约60 mL,静置2 h,室温超声30 min,转移并定容至100 mL,摇匀,溶液离心10 min(13 000 r·min-1)。取上清液适量,经0.22 μm的微孔滤膜滤过,取续滤液作为供试品溶液。同时测定样品水分。
2.2.1.2 干燥样品供试品溶液的制备 取样品粉末0.5 g,精密称定,置于具塞锥形瓶,加50%乙醇约60 mL,静置2 h,室温超声30 min,转移并定容至100 mL,摇匀,离心10 min(13 000 r·min-1)。取上清液适量,经0.22 μm的微孔滤膜滤过后,取续滤液作为供试品溶液。同时测定样品水分。
2.2.2 色谱分析条件 色谱柱:ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 μm)。流动相:A-乙腈,B-0.1%甲酸水溶液,梯度洗脱(0~2 min,3%~3% A;2~6 min,3%~10% A;6~9 min,10%~15% A;9~12 min,15%~15% A;12~16 min,15%~25% A)。流速:0.4 mL·min-1,柱温:35 ℃。
2.2.3 标准曲线的制备 精密称取干燥至恒重的酚性甘油酯对照品适量,加甲醇制成混合对照品储备液。取不同体积的上述储备液加甲醇稀释后,制成不同浓度的对照品溶液,按照2.2.2项色谱条件,注入UPLC仪进行测定,各成分线性范围分别为:Fer,13.70~137.0 μg·mL-1(r=0.999 8);Cou,15.00~150.0 μg·mL-1(r=0.999 5);Reg D,1.65~16.50 μg·mL-1(r=0.999 8);Reg C,10.00~100.0 μg·mL-1(r=0.999 7);Reg A,20.30~203.0 μg·mL-1(r=0.999 9);Reg E,1.16~11.60 μg·mL-1(r=0.999 9)。
2.2.4 样品测定 分别精密吸取对照品溶液与供试品溶液各3 μL,注入UPLC仪测定,总酚性甘油酯含量为各酚性甘油酯含量之和。结果见表3、4。
2.3 核苷测定[7-9]
2.3.1 供试品溶液的制备
2.3.1.1 烫片样品供试品溶液的制备 取样品约3 g,精密称定,加适量60%乙醇,匀浆,置于具塞锥形瓶,加60%乙醇约40 mL,静置30 min,热提(70 ℃)1 h,待溶液冷却至室温,转移并定容至50 mL,摇匀,溶液离心10 min(13 000 r·min-1)。取上清液适量,经0.22 μm的微孔滤膜滤过,取续滤液作为供试品溶液。同时测定样品水分。
2.3.1.2 干燥样品供试品溶液的制备 取0.5 g样品粉末,精密称定,置于具塞锥形瓶,加60%乙醇约40 mL,静置30 min,热提(70 ℃)1 h,待溶液冷却至室温,转移并定容至50 mL,摇匀,溶液离心10 min(13 000 r·min-1)。取上清液适量,经0.22 μm的微孔滤膜滤过,取续滤液作为供试品溶液。同时测定样品水分。
2.3.2 色谱分析条件 色谱柱:ACQUITY UPLC BEH Amide色谱柱(100 mm×2.1 mm,1.7 μm)。流动相:A-含10 mmol·L-1醋酸胺和0.8%醋酸的水溶液,B-含0.05%醋酸乙腈溶液,梯度洗脱(0~6 min,10%~10% A;6~8 min,10%~40% A;8~9 min,40%~40% A;9~11 min,40%~50% A)。流速:0.4 mL·min-1,柱温:35 ℃。
2.3.3 质谱检测条件 离子化模式:ESI+;检测方式:多反应检测(MRM);毛细管电压:3.0 kV;离子源温度:150 ℃;脱溶剂气温度:550 ℃;脱溶剂气流量:1 000 L·h-1;锥孔气流量:50 L·h-1;碰撞气流量:0.15 mL·min-1。
2.3.4 标准曲线的制备 精密称取核苷对照品适量,加10%甲醇制成混合对照品储备液。取不同体积的上述储备液加10%甲醇稀释后,制成不同浓度的对照品溶液,按照2.3.2项色谱条件,注入UPLC仪进行测定,各成分线性范围分别为:Gua,0.18~17.80 μg·mL-1(r=0.999 6);Xan,0.09~9.20 μg·mL-1(r=0.999 9);Thym,0.13~13.40 μg·mL-1r=0.999 5);Ade,0.12~12.30 μg·mL-1(r=0.999 6);Ino,0.14~14.30 μg·mL-1(r=0.999 8);Camp,0.08~8.20 μg·mL-1(r=0.999 4);Cytd,0.14~13.90 μg·mL-1(r=0.999 1);2′B,0.10~10.00 μg·mL-1(r=0.999 6);2′X,0.17~16.50 μg·mL-1(r=0.999 8);2′Niao,0.14~14.00 μg·mL-1(r=0.999 2);2′N,0.11~10.90 μg·mL-1(r=0.999 2);Thyd,0.13~13.30 μg·mL-1(r=0.999 9)。
2.3.5 样品测定 分别精密吸取对照品溶液与供试品溶液各1 μL,注入UPLC仪测定,总核苷含量为各核苷含量之和。结果见表3、4。
2.4 氨基酸的测定[10]
2.4.1 供试品溶液的制备 样品供试品溶液制备方法参照2.3.1项。
2.4.2 色谱分析条件 色谱柱:ACQUITY UPLC BEH Amide色谱柱(100 mm×2.1 mm,1.7 μm)。流动相:A-含5 mmol·L-1甲酸胺、5 mmol·L-1醋酸胺和0.2%甲酸水溶液,B-含1 mmol·L-1甲酸胺、1 mmol·L-1醋酸胺和0.2%甲酸乙腈溶液,梯度洗脱(0~6 min,15%~20% A;6~10 min,20%~30% A;10~11 min,30%~46% A)。流速:0.4 mL·min-1,柱温:35 ℃。
2.4.3 质谱检测条件 参照2.3.3项。
2.4.4 标准曲线的制备 精密称取氨基酸对照品适量,加水制成混合对照品储备液。取不同体积的上述储备液加水稀释后,制成不同浓度的对照品溶液,按照2.4.2项色谱条件,注入UPLC仪进行测定,各成分线性范围分别为:Gly,0.28~27.60 μg·mL-1(r=0.999 1);GABA,0.26~26.40 μg·mL-1(r=0.999 6);Leu,0.21~21.20 μg·mL-1(r=0.999 8);Met,0.25~25.20 μg·mL-1(r=0.999 6);Phe,0.15~15.20 μg·mL-1(r=0.999 4);Trp,0.21~20.80 μg·mL-1(r=0.999 5);Ala,0.22~22.40 μg·mL-1(r=0.999 8);Thr,0.22~22.00 μg·mL-1(r=0.999 9);Ser,0.27~27.20 μg·mL-1(r=0.999 9);Asn,0.26~26.00 μg·mL-1(r=0.999 7);Gln,0.17~16.80 μg·mL-1(r=0.999 7);Glu,0.28~27.60 μg·mL-1(r=0.999 6);Cit,0.16~15.60 μg·mL-1(r=0.999 6);Pro,0.26~26.40 μg·mL-1(r=0.999 6);Val,0.15~15.20 μg·mL-1(r=0.999 5);Tyr,0.09~9.20 μg·mL-1(r=0.999 5);Hpro,0.13~12.80 μg·mL-1(r=0.999 9);Ile,0.23~22.80 μg·mL-1(r=0.999 9);Hit,0.28~27.60 μg·mL-1(r=0.999 9);Arg,0.16~15.60 μg·mL-1(r=0.999 9)。
2.4.5 样品测定 分别精密吸取对照品溶液与供试品溶液各1 μL,注入UPLC仪测定,总氨基酸含量为各氨基酸含量之和。结果见表3、4。
2.5 多糖测定[11]
2.5.1 供试品溶液的制备
2.5.1.1 烫片样品供试品溶液的制备 取样品约3 g,精密称定,加适量80%乙醇,匀浆,置于具塞锥形瓶,加80%乙醇约40 mL,静置2 h,室温超声30 min。离心10 min(3 000 r· min-1),弃上清液,沉淀以少量80%乙醇洗涤2次。沉淀转移至圆底烧瓶中,加水约80 mL,100 ℃回流2 h,待溶液冷却至室温,转移并定容至100 mL,摇匀,溶液离心10 min(13 000 r·min-1)。精密量取上清液1 mL,以水定容至10 mL,摇匀即得。同时测定样品水分。
2.5.1.2 干燥样品供试品溶液的制备 取样品粉末(40目)约0.5 g,精密称定,加80%乙醇约40 mL,静置2 h,室温超声30 min。离心10 min(3 000 r·min-1),弃上清液,沉淀以少量80%乙醇洗涤2次。沉淀转移至圆底烧瓶中,加水约40 mL,100 ℃回流2 h,待溶液冷却至室温,转移并定容至100 mL,摇匀,溶液离心10 min(13 000 r·min-1)。精密量取上清液1 mL,以水定容至10 mL,摇匀即得。同时测定样品水分。
2.5.2 标准曲线的制备 精密称取经105 ℃干燥至恒重的无水葡萄糖对照品适量,置量瓶中,加水制成葡萄糖对照品储备液。精密量取上述储备液0.1,0.3,0.5,0.7,0.9,1.0 mL,分别置10 mL具塞试管中,加水至1.0 mL,摇匀,精密加入5%苯酚溶液2.0 mL,混匀,精密加入浓硫酸7.0 mL,混匀,置沸水浴中加热20 min,水浴冷却5 min后,于488 nm处测吸光度。准确量取水1.0 mL,自“精密加入5%苯酚”起,同法操作,作空白对照。线性范围为20.10~201.0 μg·mL-1(r=0.998 7)。
2.5.3 样品测定 取供试品溶液0.5 mL,按照2.5.2项“加水至1.0 mL,……于488 nm处测吸光度”测定多糖含量,结果见表3、4。

表3 百合烫片样品含量测定(n=3) /mg·g-1
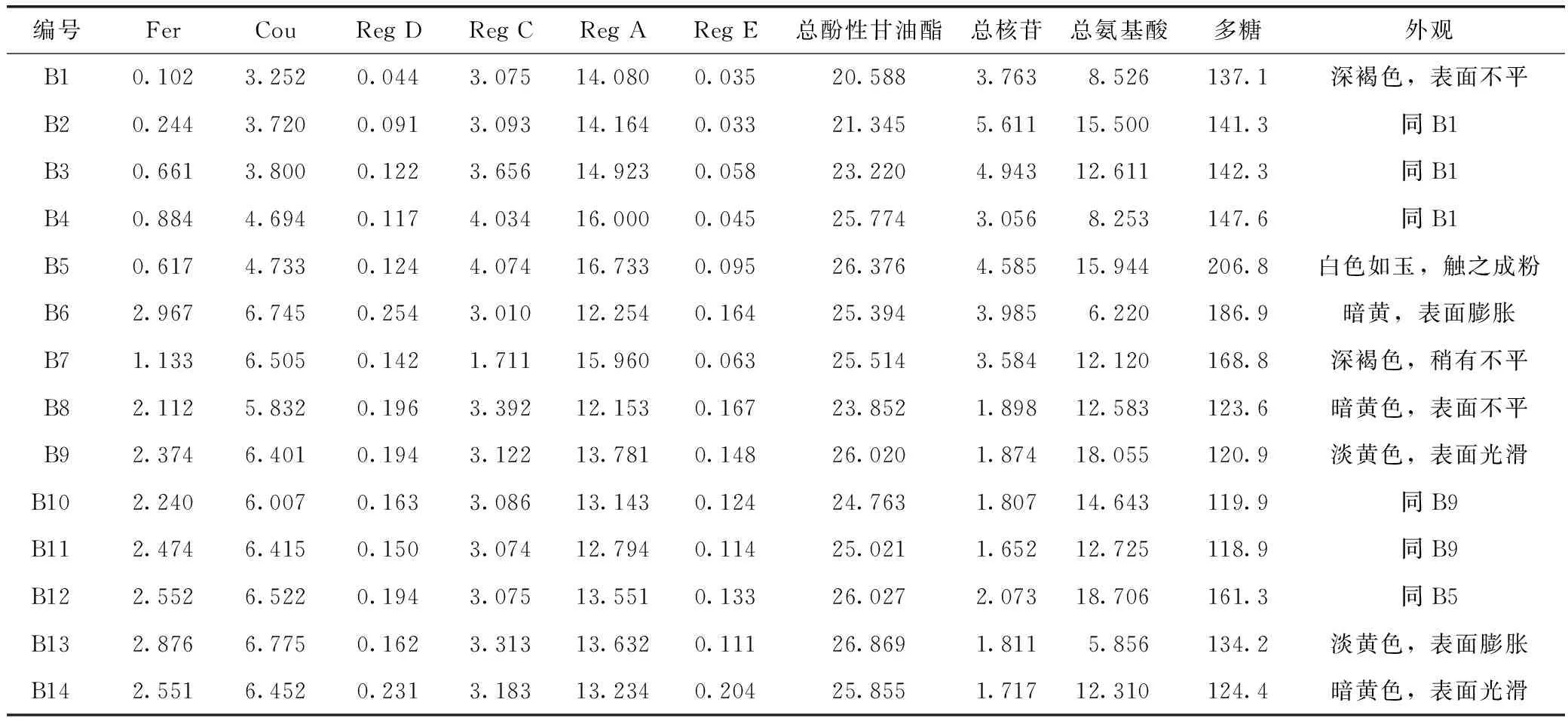
表4 百合干燥样品含量测定(n=3) /mg·g-1
3 讨论
3.1 测定方法学考察
对实验中各种检测方法均进行线性、精密度、稳定性、重复性及加样回收率考察,结果表明各指标均符合含量测定要求,适于检测相应成分。
3.2 烫片过程对百合质量的影响
煮制及蒸制样品中酚性甘油酯含量与鲜品相比几无变化,表明百合中酚性甘油酯在烫片过程得到很好保留。但烫片过程中多糖、核苷及氨基酸含量与鲜品相比均有不同程度的减少,表明水溶性成分在烫制过程中会有一定的损失,且损耗量随烫制时间加长而增加。煮5~8 min和蒸8~10 min的样品不仅外观色泽较佳,而且各成分的含量较高。因煮制操作方便易行,认为煮制5~8 min为较好的烫片工艺。
3.3 干燥过程对百合质量的影响
根据烫片工艺的优化结果,选择煮8 min的样品作为干燥工艺优化的实验对象。比较新鲜样品直接干燥与烫制样品干燥后外观,结果显示煮制品干燥后外观色泽较佳,而鲜品干燥的样品外观色泽较差,灰暗至深褐色,易出现焦糊状;烫片样品经干燥其酚性甘油酯类成分含量均略高于鲜品直接干燥。原因是未经烫片的鲜品在干燥过程中会发生酶促褐变,导致色泽的改变和营养成分的损失[12],其酚性甘油酯类成分含量减少可能是因为新鲜百合中含有破坏酚羟基的多酚氧化酶,也可能是存在破坏酯基的羧酸酯酶,烫片过程则经过短时间高温起到抑制百合内活性酶的作用,而阻止一系列生化反应[13-14],鲜品冷冻干燥样品在外观和酚性甘油酯类成分含量与经烫片干燥样品基本相近,表明冷冻干燥在较低的温度下,也可使酶活性降低。因此,烫片是百合干加工过程中必不可少的环节。干燥温度也会影响百合的质量,温度过高或过低都会使其质量降低,温度控制在60~80 ℃适宜。
3.4 适宜的百合干生产工艺
根据研究结果,确定烫片和干燥过程工艺参数为:煮5~8 min,60~80 ℃烘干或冷冻干燥。但冷冻干燥法能耗高、生产成本大,产地加工时应酌情考虑。
[1] 国家药典委员会.中国药典[S].一部.北京:中国医药科技出版社,2010:123.
[2] 中国科学院中国植物志编委会.中国植物志[M].第14卷.北京:科学出版社,1980:159.
[3] 黄燕萍.百合的研究现状[J].中国药业,2010,19(8):88.
[4] 焦力,商万有.百合加工价值研究[J].吉林农业,2011,(10):199.
[5] 胡文彦,段金廒,钱大玮,等.卷丹化学成分研究[J].中国中药杂志,2007,16(32):1656.
[6] 胡文彦,段金廒,钱大玮,等.反相高效液相色谱法测定百合中两种酚酸甘油苷的含量[J].时珍国医国药,2007,18(1):27.
[7] 曹琰,严辉,段金廒,等.不同产地当归药材核苷类成分的分析[J].药物分析杂志,2011,(11):2026.
[8] 孔德平,钱大玮,郭盛,等.9种果实、种子类补益中药的核苷类成分分析[J].中国实验方剂学杂志,2011,17(4):98.
[9] Tao W W,Duan J A,Yang N Y,et al.Determination of nucleosides and nucleobases in the pollen ofTyphaangustifoliaby UPLC-PDA-MS[J].Phytochem Anal,2012,23:373.
[10] 袁晓环,杨旭东,王春涛,等.水蛭提取液中的氨基酸的HPLC测定[J].中国医药工业杂志,2007,38(8):590.
[11] 李勇,姚曦.不同炮制方法对佛手总多糖含量的影响[J].中国药业,2012,21(4):24.
[12] 华平,郑艺梅,刘海波.不同干燥方法对百合品质的影响[J].安徽农业科学,2004,32(2):312.
[13] 滕霞,孙曼霁.羧酸酯酶研究进展[J].生命科学,2003,15(1):31.
[14] 梁迎暖,郭巧生,张重义,等.不同加工方法对怀菊品质的影响[J].中国中药杂志,2007,32(21):2313.
StudyontheEffectofDifferentProcessingMethodsinLiliumlancifoliumThunb.Bulbs
NIE Hui1, 2*, YAN Hui2, QIAN Da-wei2*, DUAN Jin-ao2, OUyang Zhen1, QIAN Ye-fei2, GUAN Han-liang2
(1.SchoolofPharmacy,JiangsuUniversity,Zhenjiang212013,China;2.JiangsuKeyLaboratoryforHighTechnologyResearchofTraditionalChineseMedicineFormulae,NanjingUniversityofChineseMedicine,Nanjing, 210046,China)
The purpose of this paper is to give support to establishing a standard processing method forLiliumlancifoliumThunb. bulbs, by comparing the changes on chemical compositions via different processes. Steam and boiling water was used in processing fresh bulbs. And 4 kinds of drying methods were used to deal with the boiled samples, including heated air drying, microwave drying, far infrared drying and freeze drying. Ultra-high performance liquid chromatography along with a diode array detector (UPLC-DAD) was used for analyzing phenylpropenoid glycosides, ultra-high performance liquid chromatography combined with a triple quadrupole electrospray tandem mass spectrometry (UPLC-TQ/MS) was applied for measuring nucleosides and amino acids, UV spectrophotometry (UV) was used for determining polysaccharide, respectively. It was in good appearance and higher contents that the samples were steamed 8~10 min or boiled 5~8 min, heated air dried at 60~80 ℃ and freeze dried. It was concluded that the boiling process was absolutely necessary for the primary processing. With considering various factors, the optimal conditions were obtained as following: the boiling time was 5~8 minutes, and the drying method was heated air drying at 60~80 ℃ or freeze drying.
LiliumlancifoliumThunb.; Boiling processing; Drying methods; Technological optimization
2013-01-10)
△
国家中医药管理局行业科研专项(201107009)
*
钱大玮,Tel:(025)85811916,E-mail:qiandwnj@126.com
猜你喜欢
航天电子对抗(2022年4期)2022-10-24 13:38:30
学生天地(2020年19期)2020-06-01 02:11:30
中国科技纵横(2018年2期)2018-11-29 18:45:44
人大建设(2018年6期)2018-11-17 22:51:26
中成药(2018年7期)2018-08-04 06:04:26
传记文学(2017年4期)2017-04-25 23:09:32
中国塑料(2016年2期)2016-06-15 20:30:00
传奇故事(破茧成蝶)(2015年1期)2015-02-28 09:26:32
食品工业科技(2014年15期)2014-03-11 18:17:45
湖南农业科学(2014年3期)2014-02-27 14:28:21
