
Error-prone PCR致哈茨木霉几丁质酶突变的条件优化
2013-09-20 13:24:50王傲雪陈秀玲李景富
东北农业大学学报 2013年10期
王傲雪,李 微,陈秀玲,李景富
(1.东北农业大学园艺学院,哈尔滨 150030;2.东北农业大学生命科学学院,哈尔滨 150030)
番茄真菌性病害已成为影响番茄生产的主要病害,严重影响番茄品质和产量[1]。哈茨木霉(Trichnderma harzianum)是一种广泛应用于植物真菌病害防治的重寄生真菌[2],能够通过拮抗作用防治番茄灰霉病[3]。哈茨木霉菌株产生的包括几丁质酶在内的细胞壁降解酶[4],在抑菌中起重要作用。在植物病害防治中,几丁质酶在有效防治植物病原真菌的同时,还可以刺激植物分泌几丁质酶,从而增强植物对病原菌的抵抗力,因而在生物防治中具有广阔的应用前景。
Error-prone PCR原理是利用DNA聚合酶不具备3'→5'校对性质,在PCR过程中出现碱基错配从而进行特定基因随机诱变的技术。此方法可获得改善活性和稳定性的突变产物,因此易错PCR以其操作简便、有效等优点成为目前应用最为广泛的进化手段之一[5-6]。Nakaniwa等运用Errorprone PCR方法对一种耐热性果胶酸裂解酶(PL47)进行体外定向进化,得到3个低耐热的突变株,突变株在耐热性和耐热活力上均低于PL47[7]。Deng等运用Error-prone PCR方法对Escherichia coli氨基葡萄糖合成酶(GlmS)进行改良,得到10个突变株,其中一个突变株蛋白酶的活性为野生型的2倍[8]。Niu等通过DNA shuffling和易错PCR的方法对根霉菌脂肪酶进行定向改造,从而得到热稳定性提高的突变株,并证明对酶热稳定性和最适作用温度起决定性作用的位于190位点[9]。
本试验通过调整Error-prone PCR的试验条件:提高Mg2+浓度,加入Mn2+,调整dNTP比例等因素,得到使哈茨木霉几丁质酶基因产生突变的的试验条件,为后期对哈茨木霉几丁质酶进行体外定向进化,获得酶活大幅提高的突变酶奠定坚实基础。
1 材料与方法
1.1 材料
1.1.1 菌种
哈茨木霉(Trichoderma harzianum)、大肠杆菌(Escherichia coli)DH5α由东北农业大学番茄研究所保存。
1.1.2 供试药剂
RNA提取试剂Trizol试剂购自Invitrogen公司;TaqDNA聚合酶、MgCl2、PCR Buffer购自北京全式金生物技术有限公司;dNTPs购自ToYoBo公司;RevertAidTM First Strand cDNA Synthesis Kit购自Fermentas公司;DNA Marker、pMD18-T Vector试剂盒购自TaKaRa公司;胶回收试剂盒、PCR产物纯化试剂盒(上海生物工程有限公司);其他试剂均为国产,分析纯。
1.1.3 供试培养基
LB培养基:用于大肠杆菌及大肠杆菌转化子(加Amp后使用,每50 mL培养基加50 μL Amp)的培养、筛选。10 g·L-1胰蛋白胨,5 g·L-1酵母提取物,5 g·L-1NaCl,15 g·L-1琼脂(Agar)配制固体培养基。1.05 kg·cm-2、121℃条件下灭菌20 min。
1.2 方法
1.2.1 哈茨木霉几丁质酶Chit42基因的诱导表达
8是一个吉利的数字,在中国人的传统语境中,8意味着财富的增长。但是在这个尾数是8的年份里,很多民众的感受中,这并不是一个好的年景。中国股市复制了2008年的大跌模式,股指在2018年开市的最后一天回落至2500点以下收盘,全年跌幅超过四分之一,1.46亿股民人均亏损近10万元。“二八规则”也被轻易打破,仅有一成股民侥幸赚了点钱。对于中国股市,这显然是一个没能逾越的寒冬。
将哈茨木霉(Trichoderma harzianum)接种到PDA培养基上活化,25℃恒温箱中培养5~8 d,直至产生大量绿色孢子,5 mL灭菌水浸泡3~5 min,轻刮洗下分生孢子,按8%的接种量接种至装有50 mL产酶发酵培养基和PD培养基的250 mL三角瓶中,置于恒温摇床,25℃,180 r·min-1培养5 d。
1.2.2 哈茨木霉总RNA的提取
将0.1 g菌丝置于研钵中,加入液氮充分研磨,迅速加入1.0 mL Trizol,剧烈震荡混匀,以剪切基因组DNA;加入200 μL氯仿/异戊醇(24∶1),剧烈振荡混匀30 s;4 ℃,12 000 r·min-1,离心5 min;将含有RNA的上清液小心转移到RNase-free 1.5 mL离心管中,加入等体积的异丙醇,室温放置5 min;4 ℃,12 000 r·min-1,离心5 min,小心移去上清液,防止RNA沉淀丢失;用75%乙醇(DEPC水配制)洗涤两次,每次700 μL,12 000 r·min-1,4 ℃离心2 min,尽可能彻底吸走上清,防止RNA沉淀丢失。真空离心干燥3~5 min,或放在室温下使酒精完全挥发;沉淀用50 μL DEPC-H2O溶解。如发现沉淀难溶,68℃处理10 min。吸取3~5 μL RNA溶液用于下步检测反应,其余RNA溶液贮存于-70℃备用。
1.2.3 哈茨木霉总RNA的质量检验
对制备的RNA进行1%琼脂糖凝胶电泳检测,紫外分光光度计检测A260/A280比值。
1.2.4 反转录为cDNA的体系设定及反应条件
反转录体系:总 RNA 3 μL(约 1 μg),Oligo(dT)引物 1 μL,ddH2O 9 μL,混匀,稍离心,70℃,5 min,立即置于冰上,冷却2 min,稍混匀离心,按顺序加入下列物质:5×Buffer 5 μL,dNTP Mixture(10 mmol·L-1) 0.5 μL,RNA酶抑制剂(25 U)1 μL,MMLV 1 μL,混匀稍离心,42 ℃反应90 min。反应完成后,存于-70℃备用。
1.2.5 常规PCR扩增
Chit42H:5'CCAAGCTTACCTCTACCAACATC ACAAGCAA 3'
Chit42N:5'CGGCGGCCGCCAGCCTAGTTCAG ACCATTC 3'
以哈茨木霉cDNA为模板进行PCR扩增,每50 μL 体系:10×Taq Buffer(含 Mg2+)5 μL;dNTP(2.0 mmol·L-1)5 μL,引物各 1 μL(0.5 μmol·L-1),模板cDNA 2 μL,Easy-Taq 0.5 μL,ddH2O 35.5 μL。几丁质酶基因Chi42扩增的反应条件为:94℃,2 min;94℃,30 s;58℃,30 s;72℃,1 min,共35个循环;反应结束后72℃延伸10 min。
1.2.6 Error-prone PCR扩增
以克隆的哈茨木霉几丁质酶基因为模板,在常规PCR基础上,尝试以下反应条件以及条件组合:①MnCl2浓度分别为:0、0.2、0.4、0.5、0.6 mmol·L-1;② MgCl2浓度分别为:1.5、2.5、5.0、6.0、7.0 mmol·L-1;③ dATP,dGTP 浓度为 0.04 mmol·L-1;dCTP,dTTP 终浓度梯度为 0.2、0.5、1.0、1.5、2.0 mmol·L-1;④不同突变条件的联合应用。
根据以上试验结果,为获得适宜的突变率,将影响突变率的因素组合。保持其他组分和扩增条件不变,在 5.0 mmol·L-1Mg2+、0.5 mmol·L-1Mn2+、1.5 mmol·L-1dCTP和dTTP条件下进行易错PCR扩增。
1.2.7 Error-prone PCR引入突变的鉴定
琼脂糖电泳分离易错PCR产物,切下所需条带,电洗脱回收,使用pMD18-T载体系统,按厂家提供的操作程序克隆到pMD18-T中,随机挑选克隆,送华大基因公司测序。
2 结果与分析
2.1 RT-PCR扩增Chit42基因
2.1.1 哈茨木霉总RNA的提取
由图1可知,RNA由3条谱带组成:分别是28SrRNA、18SrRNA和5SrRNA。并且28SrRNA与18SrRNA条带亮度比大于2∶1。紫外分光光度检测A260/A280比值为1.95,说明总RNA质量较好,可以用于后续试验。
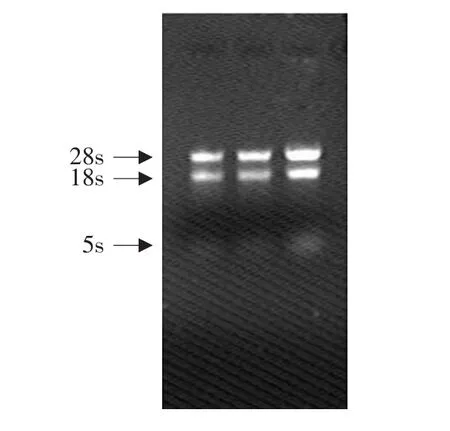
图1 哈茨木霉总RNAFig.1 Total RNA electrophoresis results of Trichoderma harzianum
2.1.2 RT-PCR扩增Chit42基因
以哈茨木霉cDNA为模板,进行PCR反应,1%琼脂糖凝胶电泳进行检测(见图2)。结果表明扩增得到1 300 bp左右的片段,与预期条带大小相符。将此片段回收,回收产物连接pMD18-T载体,转化大肠杆菌(Escherichia coli)DH5α感受态细胞,挑取阳性克隆,经菌落PCR以及酶切鉴定后,送菌液测序。测序结果经Blast比对,与哈茨木霉几丁质酶Chit42 cDNA(S78423)序列同源性达100%。

图2 RT-PCR产物电泳结果分析Fig.2 Electrophoresis analysis of RT-PCR amplification products
2.2 Error-prone PCR条件的确定
2.2.1 Mg2+浓度条件的确定
Mg2+的作用主要是dNTP-Mg与核酸骨架相互作用,增加Mg2+浓度能够稳定非互补的碱基对并能影响DNA聚合酶的活性,因此高浓度Mg2+可增加PCR非特异性扩增的效率,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而扩增不出条带。不同Mg2+浓度下PCR扩增结果见图3,可见浓度1.5、2.5、5.0 mmol·L-1均能扩增出目的片段,浓度为6.0、7.0 mmol·L-1无条带检出。

图3 不同Mg2+浓度下的Error-prone PCRFig.3 Error-prone PCR results conducted with different Mg2+concentration
2.2.2 Mn2+浓度条件的确定
Mn2+在PCR中通常作为替代Mg2+的天然辅助因子,MnCl2能够降低聚合酶对模板的特异性,因此Mn2+的加入更有助于突变的发生。不同Mn2+浓度下PCR扩增结果见图4,可见浓度为0.6 mmol·L-1时,基本得不到扩增产物,因此控制Mn2+的最高浓度为0.5 mmol·L-1。
2.2.3 dNTP浓度条件的确定
在Error-prone PCR中,将dATP、dGTP降至常规PCR浓度的20%,增加某些dNTP的浓度能够促进错误掺入。不同dCTP、dTTP浓度下PCR扩增结果见图5。可见dCTP、dTTP浓度2.0 mmol·L-1时无目的条带检出,因此控制dNTP各浓度配比为:dATP、dGTP 0.04 mmol·L-1;dCTP、dTTP最大浓度控制在1.5 mmol·L-1。

图5 不同dNTP配比下的Error-prone PCRFig.5 Error-prone PCR conducted with different dNTP concentration
2.2.4 不同突变条件的联合应用
根据离子浓度、dNTP配比的试验结果,为获得适宜的突变率,将影响突变率的因素组合。保持Error-prone PCR的其他组分和扩增条件不变,在5.0 mmol·L-1Mg2+,0.5 mmol·L-1Mn2+,0.04 mmol·L-1dATP和dGTP,1.5 mmol·L-1dTTP和dCTP条件下进行扩增。扩增结果见图6,可见在该条件下扩增良好。
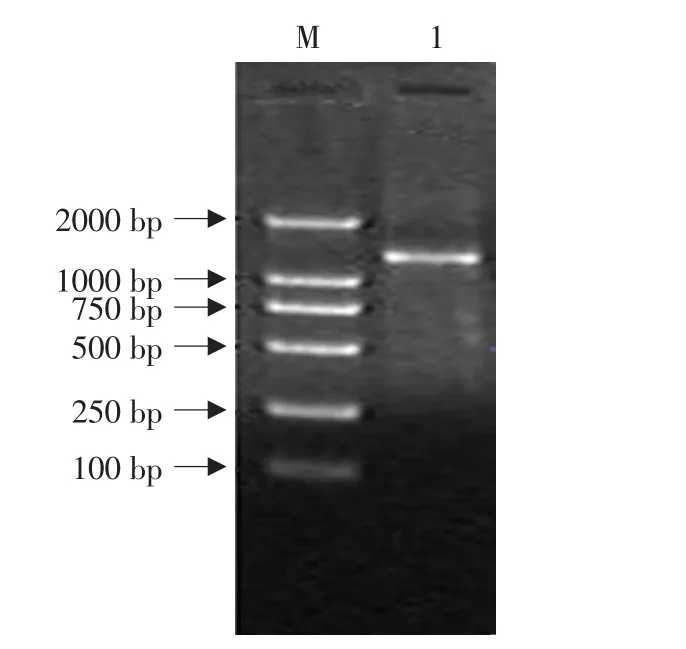
图6 Error-prone PCR产物电泳结果分析Fig.6 Electrophoresis analysis of Error-prone PCR products
2.2.5 Error-prone PCR致Chit42突变结果
共测定10个克隆,累积13 370个碱基,检测到57个突变,平均突变率为0.4%(见表1)。说明浓度为5.0 mmol·L-1Mg2+和0.5 mmol·L-1Mn2+,配合浓度为 0.04 mmol·L-1dATP,dGTP;1.5 mmol·L-1dTTP和dCTP,能够有效的进行Error-prone PCR扩增,实现碱基的突变。
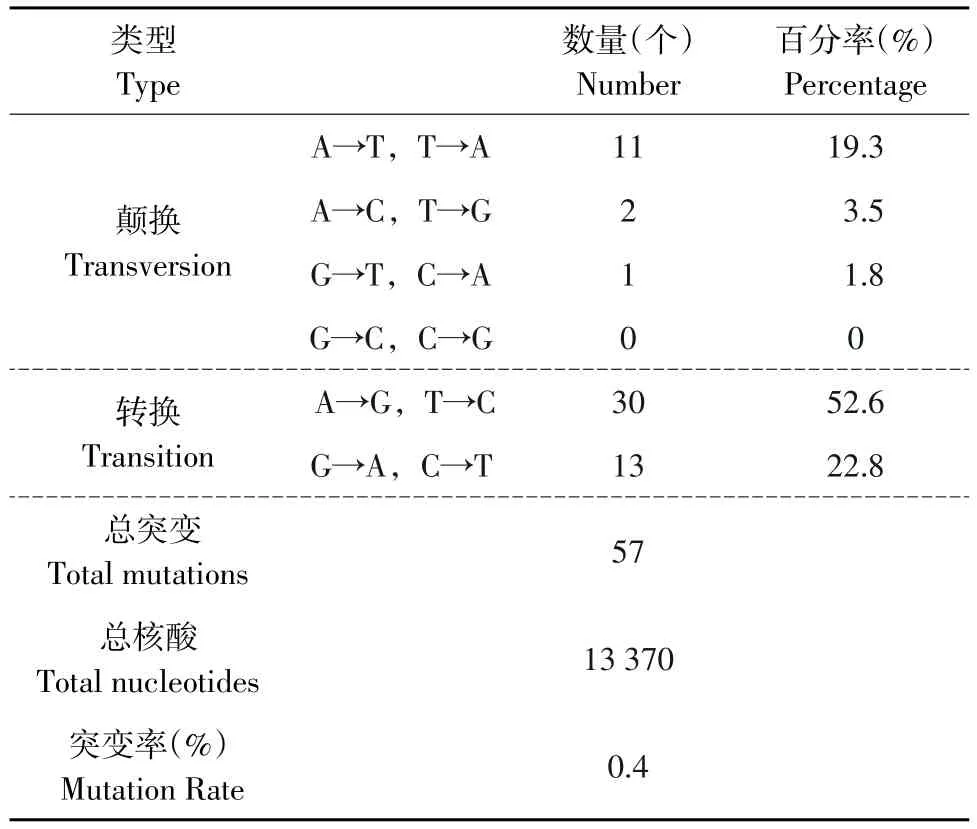
表1 错配PCR形成随机突变的类型Table1 Types of random mutation introduced by Error-prone PCR
3 讨论
随着基因工程发展及生物信息学对蛋白质功能分析深入,已开始在分子水平上对酶进行定向进化[10]。易错PCR进行随机突变简便易行,容易引入突变,突变频率高于传统的化学和物理诱变[11],是目前实验室常用的蛋白质分子改造方法。一般易错PCR产生的突变率范围为0.2%~2%。结果共测定10个克隆,累积13 370个碱基,检测到57个突变,均为点突变,未出现缺失和插入性突变,平均突变率为0.4%。发生于A/T的突变占75.4%(43/57),其中转换(A→G,T→C)占52.6%,颠换(A→T,T→A;A→C,T→G)占 22.8%(A→T,T→A:19.3%;A→C,T→G:3.5%)。而发生于G/C的突变只占24.6%(14/57),其中转换(G→A,C→T)占22.8%,颠换(G→T,C→A)占1.8%,CG→GC的颠换突变未见产生。突变后生成A/T的占43.9%(25/57),生成C/G的占56.1%(32/57)。碱基发生转换的比例远大于颠换,表明在易错PCR过程中,碱基突变具有一定偏向性。可见在Errorprone PCR中,提高Mg2+浓度稳定非互补的碱基对;引入Mn2+降低聚合酶对模板的特异性;降低dATP,dGTP浓度,增加dCTP,dTTP浓度促进错误掺入,能够有效实现碱基突变,为后期建立突变库,筛选优良突变株、酶分子定向进化奠定基础。
[1]姚彬,王傲雪,李景富.哈茨木霉对4种番茄病原真菌抑制作用的研究[J].东北农业大学学报,2009,40(5):26-31.
[2]张丽莉,李景富,王傲雪.一株生防木霉菌株的鉴定及其对番茄保护酶的诱导作用[J].东北农业大学学报,2011,42(1):89-93.
[3]郭润芳,刘晓光,高克祥.拮抗木霉在生物防治中的应用与研究进展[J].中国生物防治,2002,18(4):180-184.
[4]李静,刘建军,赵祥颖.微生物几丁质酶的研究概况[J].山东食品发酵,2006(1):6-8.
[5]Cadwell R C,Joyce G F.Mutagenic PCR[J].PCR Methods Application,1994,3(6):136-140.
[6]Moore J C,Jin H M,Kuchner O,et al.Strategies for the in vitro evolution of protein function:Enzyme evolution by random recombination of improved sequences[J].Journal of Molecular Biology,1997,272(3):336-347.
[7]Nakaniwa T,Tada T.An in vitro evaluation of a thermostable pectate lyase by using error-prone PCR[J].Journal of Molecular Catalysis B:Enzymatic,2004,27:127-131.
[8]Deng M D,Alan D.Grund.Directed evolution and characterization of Escherichia coli glucosamine synthase[J].Biochimie,2006,88:419-429.
[9]Niu W N,Li Z P,Zhang D W.Improved thermo stability and the optimum temperature of Rhizopus arrhizus lipase by directed evolution[J].Molecular Catalysis B:Enzymatic,2006,43:33-39.
[10]陈英,朱绮霞,张搏.基于易错PCR技术的黏质沙雷氏菌脂肪酶基因LipA的定向进化[J].生物技术通报,2011(4):181-185.
[11]黄瑛,蔡勇,杨江科.基于易错PCR技术的短小芽孢杆菌YZ02脂肪酶基因BpL的定向进化[J].生物工程学报,2008,24(3):445-451.
猜你喜欢
天津农业科学(2023年2期)2023-03-14 09:03:58
智慧农业导刊(2022年22期)2022-11-17 03:36:52
上海蔬菜(2022年3期)2022-06-22 06:57:46
遗传(2021年10期)2021-11-01 10:30:08
生物技术通报(2021年4期)2021-05-14 06:01:28
中国医学创新(2020年11期)2020-06-08 10:38:37
中华肺部疾病杂志(电子版)(2020年2期)2020-05-07 00:34:24
中国实验诊断学(2020年4期)2020-04-29 14:30:28
中国果业信息(2018年6期)2018-01-20 20:04:49
中国酿造(2017年8期)2017-09-03 06:20:01
