
米诺环素对慢性脑低灌注大鼠空间学习记忆能力及海马BACE-1和Aβ表达的影响
2013-08-11 08:26李国艳余昌胤
中风与神经疾病杂志 2013年2期
李国艳, 余昌胤, 杨 华
随着我国步入老年化社会,慢性脑低灌注性认知功能障碍已成为中老年人生活质量下降的原因之一。慢性脑低灌注时有多种机制参与损伤认知功能,其病理生理过程较为复杂,主要涉及血脑屏障破环、脑白质脱髓鞘、细胞凋亡、炎症反应、胆碱能神经系统受损、Aβ产生及聚集等多个环节[1,2]。本实验拟通过结扎双侧颈内动脉建立大鼠慢性脑低灌注模型,采用米诺环素进行干预,观察米诺环素对大鼠学习记忆能力及海马区Aβ及BACE-1表达的影响,以探讨米诺环素对慢性脑低灌注大鼠学习记忆功能的影响及可能的机制。
1 材料与方法
1.1 实验动物与分组 72只SD大鼠,雌性,体重200~250g,由第三军医大学实验动物中心提供,清洁级,常规分笼饲养。采用国家标准啮齿类动物干燥饲料喂养,自由饮水进食。适应性喂养1w,然后按抓取随机原则分组:假手术组(分为造模后1个月、2个月、3个月组)、慢性脑低灌注组(模型组,分为造模后1个月、2个月、3个月组)和米诺环素治疗组(治疗组,分为造模后1个月、2个月、3个月组),每组8只。术中出血较多、出现呼吸困难及提前死亡者予剔除,并补足相应例数。
1.2 动物模型制作方法 制作慢性脑低灌注大鼠模型参照 Liu等[3]的方法:大鼠手术前禁食12h,禁水4h,用10%水合氯醛0.3ml/100g腹腔注射麻醉后仰卧固定,颈前部去毛消毒后沿颈正中切口,然后分离出双侧颈总动脉,用双重丝线行双颈总动脉永久性结扎术后,缝合皮肤。假手术对照组除不结扎颈总动脉外,其他操作与模型组相同。用生理盐水将米诺环素稀释成0.5mg/ml浓度的溶液备用。米诺环素治疗组在慢性脑低灌注模型的基础上予50mg/kg/d的米诺环素溶液灌胃;假手术组和慢性脑低灌注组每天给予等量NS灌胃。米诺环素的剂量选择参考 Stirling 等[4]和 Hewlett[5]的米诺环素神经保护作用机制研究。
1.3 Morris水迷宫实验 各组分别在造模后1个月、2个月和3个月采用Morris水迷宫对大鼠进行学习记忆能力的检测(参照Vorhees等[6]总结的方法):水迷宫为一圆形水池,其直径150cm,高60cm,池中水深32cm,水温保持22℃。在池壁上等距离分别依次标记4个方向:东(E)、南(S)、西(W)、北(N),在 ES象限(第四象限)正中离池壁30cm处放一个圆形平台,平台直径为10cm,高30cm。水池上方安有摄像头,图像经视频输入计算机,大鼠的运动轨迹被同步记录。连续训练4d,每天训练两次,每次随机在NW、W、SW和E方向中选择一个作为入水点,观察并记录大鼠爬上平台的轨迹图及所需时间(定位航行潜伏期)。如果大鼠在120s内找不到平台,则将大鼠引至平台并在平台上停留20s。第5天去除平台,选择NW作为入水点,观察并记录大鼠的游泳速度以及120s内在ES(第四象限)的游泳时间(空间探索时间),每只大鼠检测一次。
1.4 组织取材 在Morris水迷宫训练结束第2天,用10%水合氯醛0.3ml/100g于大鼠腹腔注射麻醉后,快速暴露心脏,先用生理盐水经升主动脉快速灌注冲洗3~5min,再用卵圆钳夹闭大鼠的腹主动脉,经左心室灌注4%多聚甲醛(PBS溶解,pH 7.4)约150ml,灌注固定后断头取脑,标本放入4%多聚甲醛液中浸置,24h内石蜡包埋。取海马组织连续4μm厚切片。每个标本切2张切片,本实验共144张切片。待切片干燥后72张行免疫组化染色测定海马Aβ蛋白表达,另72张行免疫组化染色测定海马BACE蛋白表达。
1.5 免疫组织化学染色法 切片脱蜡、水化,切片加入0.01mol柠檬酸钠缓冲液中微波中高火10min进行抗原修复,待冷却后,3%H2O2去离子水孵育10min,以阻断内源性过氧化物酶。PBS洗3次,每次2min。不同的切片分别加入适量兔抗人BACE多克隆抗体(1∶25稀释)或兔抗人Aβ多克隆抗体(1∶50稀释),4℃湿盒内过夜。第2天置于37℃温箱复温45min,PBS洗3次,每次2min。滴加二抗后置于37℃温箱孵育45min,DAB溶液显色5~10min,苏木精对比染色;常规脱水,封片。
1.6 图像分析 在10×40倍光镜下观察海马CA1、CA3两个分区,BACE、Aβ 阳性细胞胞浆呈棕黄色,部分细胞核亦着色。随机选取5个非重叠的视野,进行免疫反应阳性细胞数计数,以个/视野为单位,结果取5个视野的平均值。所观察的每个视野均由病理科3名高级职称人员进行阳性细胞计数,如3人判断同一视野阳性细胞数相同则纳入研究对象。图片采用Motic Image Devices 2.0图文采集系统获取。
1.7 统计学处理 采用SPSS 12.0统计学软件进行统计学处理,计量资料均采用均数±标准差()表示,数据处理组内比较采用重复测量数据的方差分析、组间比较采用单因素方差分析,P﹤0.05表示差异有统计学意义,P﹤0.01表示统计学有显著性差异。
2 结果
2.1 Morris水迷宫试验
2.1.1 在连续4d的训练中,各组大鼠每天全程训练游到平台的时间取平均值即为每天的水迷宫平均潜伏期。本试验显示,与同时点模型组比较,各假手术组和治疗组第4天定位航行潜伏期均有不同程度缩短(P <0.05,P <0.01);随着时间的推移,模型组潜伏期逐渐延长,但模型组1个月、2个月、3个月之间差异无显著性。假手术组和治疗组之间潜伏期比较差异无显著性(见表1)。
2.1.2 空间探索实验主要考察大鼠的记忆能力 第5天模型组大鼠第四象限的游泳时间(空间探索时间)明显降低,并随着时间的推移,空间探索时间逐渐缩短,模型组3个月与模型组1个月比较差异有显著性(P<0.05);与同时点模型组比较,各假手术组、治疗组空间探索时间均有不同程度增加(P <0.05,P <0.01)。假手术组和治疗组之间探索时间比较差异无显著性(见表2)。
2.2 各组大鼠脑组织BACE和Aβ的表达
2.2.1 本实验显示 假手术组和治疗组BACE的表达与同时点模型组比较明显降低(P<0.05,P<0.01)。模型组 BACE 的表达随时间逐渐增加,但组内比较差异无显著性(P>0.05)。假手术组和治疗组比较BACE的表达差异无显著性,治疗组组内比较差异无显著性。以上结果说明米诺环素能抑制BACE的表达(见表3)。
2.2.2 本实验显示 假手术组和治疗组Aβ的表达与同时点模型组比较明显降低(P<0.05,P<0.01)。模型组Aβ的表达随时间逐渐增加,其中3个月组与1个月、2个月组比较差异有显著性(P<0.05)。假手术组和治疗组比较Aβ的表达差异无显著性,治疗组组内比较差异无显著性。以上结果说明米诺环素能抑制慢性脑低灌注所致的Aβ的表达(见表4)。
表1 3组大鼠Morris水迷宫试验第4天潜伏期(s)比较(,n=8)
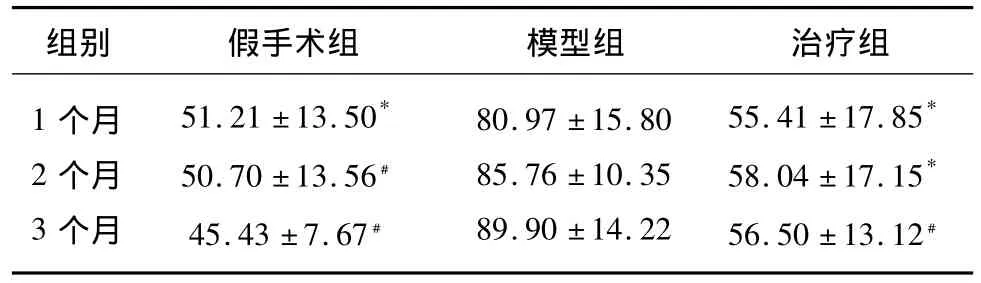
表1 3组大鼠Morris水迷宫试验第4天潜伏期(s)比较(,n=8)
与同月模型组比较*P<0.05,与同月模型组比较﹟P<0.01
组别 假手术组 模型组 治疗组1个月2个月3个月51.21 ±13.50*50.70 ±13.56﹟45.43 ±7.67﹟80.97 ±15.80 85.76 ±10.35 89.90 ±14.22 55.41 ±17.85*58.04 ±17.15*56.50 ±13.12﹟
表2 3组大鼠Morris水迷宫试验空间探索时间(s)比较()
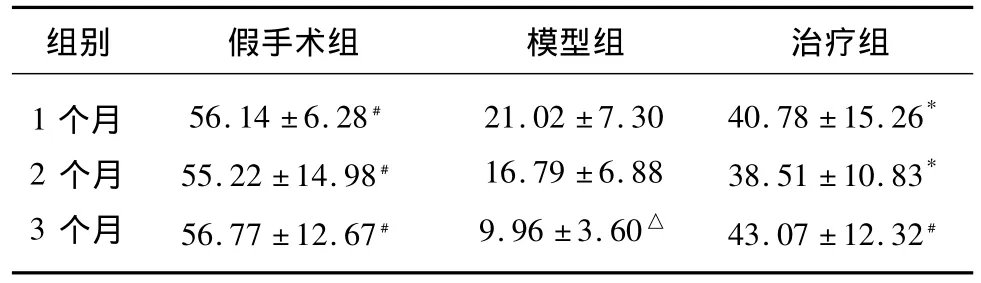
表2 3组大鼠Morris水迷宫试验空间探索时间(s)比较()
与模型组比较*P<0.05,﹟P<0.01;模型组3个月与模型组1个月比较△P<0.05
组别 假手术组 模型组 治疗组1个月2个月3个月56.14 ±6.28﹟55.22 ±14.98﹟56.77 ±12.67﹟21.02 ±7.30 16.79 ±6.88 9.96 ±3.60△40.78 ±15.26*38.51 ±10.83*43.07 ±12.32﹟
表3 3组大鼠海马BACE(个/视野)的表达()
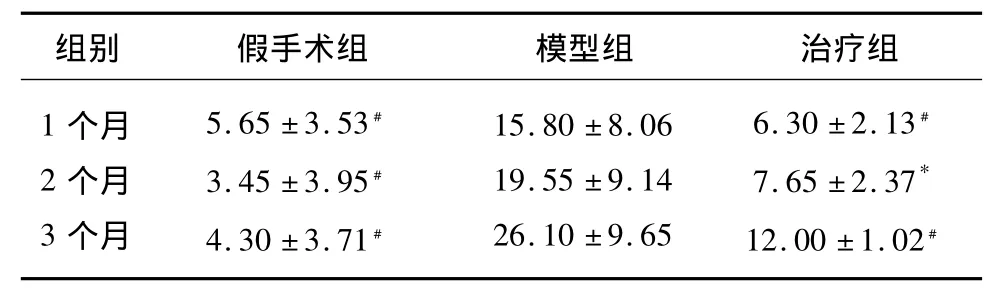
表3 3组大鼠海马BACE(个/视野)的表达()
与模型组比较*P <0.05,﹟ P <0.01
组别 假手术组 模型组 治疗组1个月2个月3个月5.65 ±3.53﹟3.45 ±3.95﹟4.30 ±3.71﹟15.80 ±8.06 19.55 ±9.14 26.10 ±9.65 6.30 ±2.13﹟7.65 ±2.37*12.00 ±1.02﹟
表4 3组大鼠海马Aβ(个/视野)的表达()
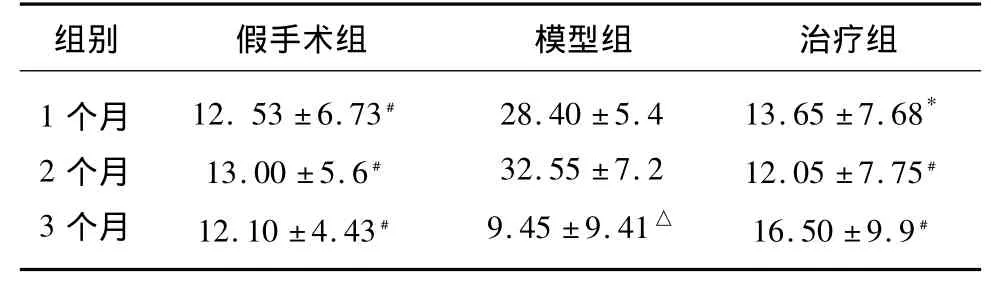
表4 3组大鼠海马Aβ(个/视野)的表达()
与模型组比较*P<0.05,﹟P<0.01;模型组3个月与模型组2个月比较△P<0.05
?
3 讨论
阿尔茨海默病(Alzheimer’s disease,AD)是一种进行性脑内神经元变性疾病,病理上主要表现为神经细胞内神经原纤维缠结(neurofibrillary tangles,NFTs)和细胞外β淀粉样蛋白(β-amyloid proteion,Aβ)沉积所致的老年斑(senile plaques,SP)的联合发生,神经原纤维缠结主要由tau蛋白过度磷酸化引起。AD病理变化中固定的一点是在大脑和边缘皮质部形成细胞外淀粉样斑块,以及在脑膜和大脑的血管壁上有淀粉样化学类似物的沉积,其主要组成是4kD的β淀粉样肽(Aβ)。42个氨基酸形成的Aβ-Aβ42的过量产生已证实是几种家族性早发性AD 共同的起因[7]。
Aβ是由其前体蛋白β淀粉样前体蛋白(β-amyloid precursor pretein,APP)水解产生的。APP有多种酶切方式,分别由不同的分泌酶参与水解过程。Aβ是β和γ分泌酶(β、γ-secretase)共同作用的结果,其中β分泌酶(β-site APP cleaving enzyme)是关键的限速酶,与AD的发生密切相关[8],对β分泌酶的研究为开发治疗AD的药物提供了依据和线索。1999年,几个独立的实验室先后报道了他们用不同的方法确定的β分泌酶,是一种具有活性的酸性蛋白水解酶,分别称为BACE-1(beta site APP cleaving enzyme)、memapsin2和Asp2,序列对比显示它们是同一分子,现在通常将其称之为BACE或BACE-1。
慢性脑低灌注(chronic cerebral hypoperfusion)是一种神经系统常见的病理状态,各种临床与基础研究显示无论是阿尔茨海默病(Alzheimer’s disease,AD)还是血管性痴呆(vascular dementia,VD)都存在程度不同的慢性脑低灌注[9]。随着生活水平的提高,慢性脑低灌注的发病呈上升趋势,慢性脑低灌注性认知功能障碍已成为中老年人生活质量下降的原因之一。
慢性脑低灌注能引起脑代谢障碍及脑功能衰退[10],有多种机制参与损伤认知功能,其病理生理过程较为复杂,其中以血脑屏障(blood-brain barrier,BBB)破坏、炎症反应和Aβ产生及聚集最为明显。AD特征性病理改变老年斑(senil plaques,SP)的主要成分是Aβ,近年的研究显示,Aβ不但存在于AD中,而且与脑缺血关系密切。基础研究显示缺血性脑血管病患者中APP和Aβ的水平较正常增加[11]。实验证明脑缺血能够使大鼠脑内 APP、β-APP RNA表达上调,使Aβ生产增加,使BACE活性和表达增加[12~14]。Torre等实验证明一过性脑缺血大鼠模型能诱导神经元BACE mRNA表达增加[15],提示脑缺血是诱导BACE活性增强的原因之一。
米诺环素(minocycline,MC)是第二代半合成的四环素类抗生素,临床上主要用来治疗感染性疾病,近年来研究表明,它不仅有抗微生物作用,还有显著的抗炎、抗凋亡和抗氧化等作用,具有较强的抗炎作用、高亲脂性、长期用药安全性较好等特点。基础研究显示四环素类药物可通过多重机制发挥其脑保护作用,包括通过抗炎机制、抑制基质金属蛋白激酶的活性、保护血脑屏障、保护神经元、清除氧自由基及抗细胞凋亡途径等。在四环素类药物中具有脑保护作用者依次为米诺环素、强力霉素和四环素。
大鼠双侧颈总动脉永久性结扎术是目前较常采用的一种慢性脑低灌注模型,结扎双侧颈总动脉后使大鼠15个脑区的局部血流量降低25%~87%。Morris水迷宫是目前主要用于检测大鼠海马相关的空间学习记忆能力的方法。通过水迷宫试验,我们的结果显示,慢性脑低灌注能使大鼠空间学习记忆能力降低,并随着时间的推移,该能力有进一步下降,特别在记忆能力方面下降更为明显,说明在慢性低灌注情况下,缺血缺氧对学习记忆功能的影响是持续的、渐进的。通过对大鼠行为学观察,本实验发现米诺环素能改善慢性脑低灌注大鼠的空间学习记忆能力。模型组定位航行潜伏期和空间探索时间与假手术对照组和米诺环素治疗组的检测结果均差异显著,米诺环素治疗组与假手术对照组检测结果无显著差异。治疗组的空间学习记忆能力较模型组明显改善,从行为学上说明了米诺环素具有脑保护作用。
APP在体内的裂解存在两种途径:非淀粉源途径和淀粉源途径[16]。非淀粉源途径(α-secretase pathway)是指在α-分泌酶作用下,在Aβ的第16位赖氨酸和17位亮氨酸之间发生裂解,因而产生分泌型α-APPs及C83;C83再在γ-分泌酶的作用下生产不完整的Aβ,称作p3成分。α-APPs具有降低细胞内Ca2+浓度、维持突触可塑性及保护神经元的功能。淀粉源途径(β-secretase pathway)是在β-分泌酶的作用下,于Aβ的氨基端裂解,生成β-APPs及C99;C99再在γ-分泌酶的作用下,于Aβ的羧基端裂解产生完整的Aβ分子。非淀粉源途径是APP代谢加工的主要途径,在病理状态下,淀粉源途径增多,Aβ的分泌量可高于正常的4~10倍,导致Aβ的异常聚集。γ-分泌酶催化位置的不同可产生由39-43个氨基酸组成的长度不同的Aβ,其中有病理意义的是 Aβ40、Aβ42,脑内 Aβ42水平远远低于 Aβ40,前者是形成老年斑的主要成分。过量的Aβ有很强的自聚性,在老化因素(如过氧化物、兴奋性氨基酸、Ca2+稳态失衡等)的作用下聚合沉淀,在神经元间形成老年斑,引发一系列神经毒性[17],最终导致AD的产生。
近年来临床研究发现,卒中后AD的发生率明显增加,显示脑缺血参与了AD的发生和发展[18]。AD患者在出现脑代谢降低、神经元变性和认知功能减退之前即存在脑低灌注,病理研究表明多数AD患者缺血性损伤和AD病理共存[19],显示血管因素所致的缺氧与AD发病过程密切相关,但具体的机制仍不清楚。有实验显示缺氧可能通过调节α分泌酶、β分泌酶(BACE)的活动(降低α分泌酶的表达、增加BACE的表达),选择性地使APP通过淀粉源途径裂解增加,从而使Aβ生产增加,促进了AD的发生与发展。
在本实验中,我们用免疫组化方法对各组大鼠海马区BACE、Aβ进行测定,结果显示模型组BACE和Aβ的表达明显高于假手术组与治疗组,表明缺血后海马Aβ表达增多,其原因可能与以下几点有关:缺血损伤激活APP基因,使APP表达增加;BACE生成增加,APP经β淀粉源途径代谢相应增多;另外缺氧导致BBB的破坏,使通过BBB清除Aβ的功能受损[20]。本实验结果显示,米诺环素能明显降低慢性脑低灌注后大鼠BACE、Aβ的表达,表明米诺环素能抑制BACE表达,使APP通过淀粉源途径裂解减少,使Aβ生成减少,从而发挥其脑保护作用。
[1]Lee JH,Park SY,Shin YW,et al.Neuroprotection by cilostazol,a phosphodiesterase type 3 inhibitor,against apoptotic white matter changes in rat after chronic cerebral hypoperfusion[J].Brain Res,2006,1082(1):182-191.
[2]Wakita H,Tomimoto H,Akiguchi I,et al.Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat[J].Brain Res,2002,924(1):63-70.
[3]Liu HX,Zhang JJ,Zheng P,et al.Altered expression of MAP-2,GAP-43,and synaptophysin in the hippocampus of rats with chronic cerebral hypoperfusion correlates with cognitive impairment[J].Brain Res Mol Brain Res,2005,139:169-177.
[4]Stirling DP,Khodarahmi K,Liu J,et al.Minocycline treatment reduces delayed oligodendrocyte death,attenuates axonal dieback,and improves functional outcome after spinal cord injury[J].J Neurosci,2004,24(9):2182-2190.
[5]Hewlett KA,Corbett D.Delayed minocycline teeatment reduces longterm functional deficits and histological injury in a rodentmodel of focal ischemia[J].Neuroscience,2006,141(1):27-33.
[6]Vorhees CV,Williams MT.Morris water maze:procedures for assessing spatial and related forms of learning and memory[J].Nat Protoc,2006,1:848-858.
[7]Citron M,Oltersdorf T,Haass C,et al.Mutation of the β-amyloid precursoe protein in familial Alzheimer’s disease increases β-protein production[J].Nature,1992,360:672-674.
[8]Haass C,Selkore DJ.Cellular processing of β-amyloid precursor protein and the genesis of amyloid β-peptide[J].Cell,1933,75:1039-1042.
[9]ladecola C.Neurovasular regulation in the normal brain and in Alzheimer’s disease[J].Nat Rev Neurosci,2004,5:347-360.
[10]Mori E.Impact of subcortical ischemic lesions on behavior and cognition[J].Ann N Y Acad Sci,2002,977:141-148.
[11]Jendroska K,Hoffmann OM,Patt S.Amyloid beta peptide and precursor protein(APP)in mild and severe brain ischemia[J].Ann N Y Acad Sci,1997,826:401.
[12]Shi J,Yang SH,Stubley L,et al.Hypoperfusion induces overexpression of β-amyloid precursor protein mRNA in a focal ischemic rodent model[J].Brain Res,2000,853(1):1-9.
[13]Wen Y,Onyewuchi O,Yang S,et al.Increased beta secretase activity and expression in rats following transient cerebral ischemia[J].Brain Res,2004,1009(1 ~2):1.
[14]Nihashi T,Inao S,Katita Y,et al.Expression and distribution of βamyloid precursor protein and β-amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion[J].Acta Neurochir(Wien),2001,143(3):287.
[15]de La Torre JC,Fortin T,Park GA,et al.Chronic cerebrovascular insufficiency induces dementia-like defieits in aged rats[J].Brain Res,1992,582(2):186.
[16]王怀明,郭洪志.阿尔茨采默病:β-淀粉样蛋白及其毒性的研究进展[J].中风与神经疾病杂志,2000,17(5):317-318.
[17]Soto C.Plaque busters:Strategies to inhibit amyloid formation in Alzheimer’s disease[J].Mol Med Today,1999:5(8):343-350.
[18]Kalaria RN.The role of cerebral ischemia in Alzheimer’s disease[J].Neurobiol Aging,2000,21:321-330.
[19]De la Torre JC.Alzheimer disease as a vascular disorder:nosological evidence[J].Stroke,2002,33:1152-1162.
[20]Lokovic BV.Neurovascular mechanisms of Alzheimer’s neurodegeneration [J].Trends Neurosci,2005,28(4):202.
猜你喜欢
垂钓(2022年2期)2022-03-01
实用老年医学(2021年7期)2021-12-04
皮肤病与性病(2021年3期)2021-07-30
当代水产(2021年3期)2021-07-20
文理导航·科普童话(2016年7期)2017-02-04
文理导航·科普童话(2016年4期)2016-05-31
儿童故事画报·智力大王(2015年12期)2016-01-23
儿童故事画报·智力大王(2015年2期)2015-05-20
癌变·畸变·突变(2015年3期)2015-02-27
青年文摘·上半月(1984年2期)1984-11-01
