
联吡啶钌配合物对末端炔烃的催化
2013-04-21 05:46:38尹传奇
武汉工程大学学报 2013年6期
尹传奇,成 军,张 平,陈 阔
(武汉工程大学化工与制药学院,绿色化工过程教育部重点实验室,湖北省新型反应器与绿色化学工艺重点实验室,湖北 武汉 430074)
0 引 言
苯及其衍生物是重要的化工原料,利用过渡金属配合物,如铑[1-2]、钯[3]、铁[4]、钴[5-6]、镍[7]、锆[8-9]和钼[10-13]的配合物等催化炔烃环三聚是制备取代苯的有效途径之一,因而配合物催化环三聚反应生成苯的衍生物成为一个热门研究方向[14-16]. 在催化炔烃的环三聚反应中,过渡金属中心原子、配体结构、炔烃的结构及反应介质等都对催化反应的产率,特别是对产物取代苯上取代基的区域选择性产生影响. 在研究含水配体的过渡金属钌配合物的催化活性时,发现其对乙腈具有催化水化作用[17]. 该类配合物对与乙腈具有相似三键结构的末端炔烃是否具有催化环三聚作用引起了笔者的极大兴趣.
1 实验部分
1.1 试剂及仪器
2-氨基-6-甲基吡啶购自武汉格奥试剂公司; 质量分数为48%的氢溴酸, 溴素, 氢氧化钠, 金属钠, 二氯甲烷均为分析纯,购自国药集团化学试剂公司;亚硝酸钠, 甲苯, 无水硫酸镁, 质量分数为36%的浓盐酸以及LiCl为分析纯,购自天津博迪化工公司; RuCl3·3H2O购自Aldrich 公司; 高纯氮气(99.999%)购自祥云气体公司.
6,6'-二甲基-2,2'-联吡啶按文献方法合成[18].甲苯用无水氯化钙预干燥后,使用前利用Schlenk技术用金属Na回流干燥(二苯甲酮为指示剂)和蒸馏脱氧. 红外光谱数据由Nicolet 420傅里叶红外光谱仪测得. 元素分析数据通过CHNOS Elemental VarioEL III元素分析仪测定. EI-MS数据由 Thermo LTQ XL型液相色谱-离子阱质谱联用仪测得. GC-MS数据由美国Agilent5975C型气-质联用仪测得.
1.2 [Cis-Ru(dmbp)2Cl2]Cl·2H2O(1)的合成
将1.50 g(5.74 mmol) RuCl3·3H2O,以及2.12 g(11.52 mmol) 6,6'-二甲基-2,2'-联吡啶和1.75 g (41.28 mmol) LiCl加入50 mL两口烧瓶中, 采用Schlenk技术在N2保护下, 加入20 mL DMF(N,N'-二甲基甲酰胺)将原料溶解, 反应液为棕黄色. 110 ℃反应18 h后, 冷却, 加入18 mL脱氧水, 溶液变成黄褐色. 过滤, 固体依次用18 mL水和8 mL丙酮洗涤, 真空干燥后得黄褐色固体2.60 g,产率74.0%. IR (KBr) ν/cm-1:3 425 (OH), 2 930 (CH3), 1 617, 1 563, 1 420(吡啶环); EI-MS (70 eV):m/z (%):540 (38) [M-Cl]+,526 (41) [M-CH3Cl]+,498 (59) [M-C3H9Cl]+,454 (83) [M-CH3Cl3]+, 438 (100) [M-C2H6Cl3]+, 427 (5) [M-C3H9Cl3]+, 413(22) [M-C4H12Cl3]+. C24H28Cl3N4O2Ru元素分析理论值: C 47.11, H 4.61 N 9.16; 实测值: C 47.13, H 4.64, N 9.12.
1.3 [Cis-Ru(dmbp)2(H2O)2] (OTf)3(2)的合成
将0.28 g (0.46 mmol) 配合物1和0.36 g (1.40 mmol) AgOTf加入到25 mL圆底烧瓶中,采用Schlenk技术在N2保护下, 加入15 mL脱氧水,搅拌下于68 ℃反应2 h,溶液变成绿色. 冷却,过滤,滤液浓缩后用少量丙酮溶解再加乙醚析出固体,过滤、干燥得绿色固体0.29 g,产率为68.0%. IR (KBr) ν/cm-1: 3 420 (OH), 1 644, 1 607, 1 468 (吡啶环), 1 246, 1 029 (磺酸盐), 870 (s, Ru—O). C27H28F9N4O11S3Ru元素分析理论值: C 50.05, H 4.20, N 9.73, S10.10; 实测值: C 50.12, H 4.25, N 9.61, S 10.22.
1.4 末端炔烃的催化环三聚反应
将95.20 mg (0.10 mmol)配合物2加入到25 mL两口烧瓶中,采用Schlenk技术在N2保护下,向烧瓶中加入5.00 mL (0.28 mol)脱氧水和5.00 mL 四氢呋喃. 再向烧瓶中加入末端炔烃 (30.00 mmol).加热回流24 h,冷却后将反应混合物用50 mL乙醚萃取,萃取液用无水硫酸镁干燥, 过滤后, 将溶剂旋干. 残留物用乙醚溶解后, 通过GC-MS检测混合物中各组分及其含量.
2 结果与讨论
2.1 配体的合成
配体6,6'-二甲基-2,2'-联吡啶依据文献方法(见图1),以2-氨基-6-甲基吡啶为原料,经氨基溴代生成2-溴-6-甲基吡啶、骨架镍催化对称偶联合成. 卤代芳香类化合物的对称偶联反应除使用骨架镍作催化剂外,还可用钯/碳催化剂[19]或NiCl2(PPh3)2[20], 但前者生产成本较高, 后者生产过程中的 PPh3有一定的毒性. 此外,用有机锂试剂2,2'-联吡啶-6,6'-二锂(6,6'-dilithio-2,2'-bipyridyl)与硫酸二甲酯反应,亦可得到配体6,6'-二甲基-2,2'-联吡啶, 但需在-90 ℃下进行[21].

图1 6,6'-二甲基-2,2'-联吡啶的合成方法Fig.1 Synthetic method of 6,6'-dimethyl-2,2'-bipyridine
2.2 配合物的合成
以RuCl3·3H2O为原料与2,2'-联吡啶反应,溶剂及反应温度对配合物的生成有非常大的影响:以乙醇为溶剂,回流12 h,得到[Cis-Ru(bpy)2Cl2]Cl·3.5H2O; 以DMF为溶剂,回流20 h,得到[Cis-Ru(bpy)2Cl2]·2H2O,DMF同时作为还原剂[22]. 生成的配合物为热力学稳定的顺式结构,其反式结构需发生光化学反应才能生成[23].
本实验采用DMF为溶剂,以RuCl3·3H2O为原料与6,6'-二甲基-2,2'-联吡啶反应合成配合物1,但反应温度控制在120 ℃,结果得到的钌配合物无1H NMR信号,说明配合物1中钌为三价.红外光谱在3 425 cm-1处显示—OH的特征峰,但无Ru—O键的特征吸收峰,在MS图谱上显示[Ru(dmbp)2Cl2]+([M-Cl]+)离子峰(540).结合元素分析数据,配合物1的分子结构为[Cis-Ru(dmbp)2Cl2]Cl·2H2O.配合物2由1与AgOTf反应脱氯制备,同样无1H NMR信号,红外光谱在870 cm-1处显示Ru—O键的特征吸收峰,因而其分子结构应为[Cis-Ru(dmbp)2(H2O)2] (OTf)3. 结果没有得到配合物1和2的晶体结构.
2.3 催化反应
配合物2对1-己炔、苯乙炔和丙炔酸乙酯的催化环三聚结果见表1. 三者中,苯乙炔的催化环三聚产率最高(64.7%),而丙炔酸乙酯的环三聚产率最低(51.5%). 环三聚产物中,没有连三苯产物检出;偏三苯与均三苯的比值,R为ph—(苯基)时最大(4.4),R为CH3(CH2)3—时最小(3.7).反应12 h时苯乙炔环三聚产率为64.2%,反应到24 h时的产率为64.7%,即反应时间为12 h时反应接近平衡.当向丙炔酸乙酯的催化反应体系中加入配体dmbp和CH3CN时,环三聚产率大大降低. 空白实验和只加入配合物1实验表明,没有配合物2的加入,就没有环三聚产物生成.

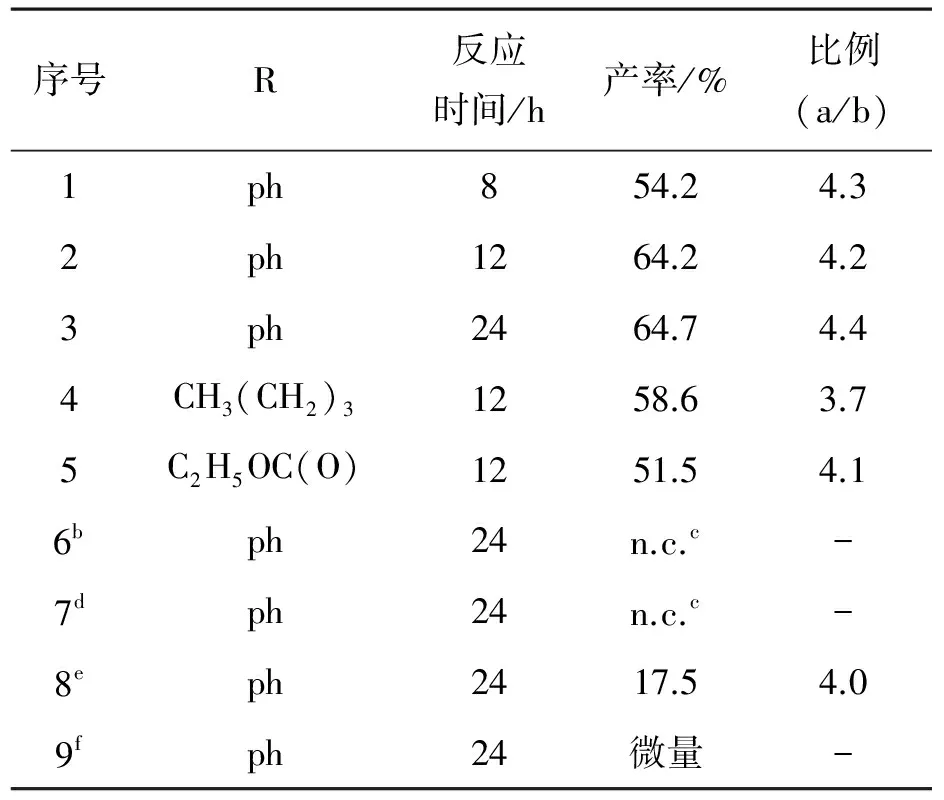
序号R反应时间/h产率/%比例(a/b)1ph854.24.32ph1264.24.23ph2464.74.44CH3(CH2)31258.63.75C2H5OC(O)1251.54.16bph24n.c.c-7dph24n.c.c-8eph2417.54.09fph24微量-
注: a 为0.10 mmol配合物2, 催化剂/底物: 1∶300 (摩尔比); 反应方式: 回流; b 为无配合物2加入; 标注c中n.c.为无环三聚产物; d 为配合物1(0.10 mmol); 标注e 为dmbp (0.50 mmol); f 为CH3CN (5 mL).
2.4 催化机理
依据文献报道[1-17,24]和实验结果,配合物2催化炔烃三聚形成取代苯的可能机理如图2所示:首先,两个水分子配体被两分子炔烃取代生成π-炔基配合物3,然后通过氧化偶联反应得到钌杂环戊二烯配合物4,第三个炔烃分子取代4中联吡啶配体上的一个配位N原子生成钌杂环戊二烯-炔基配合物5,该配合物经由插入反应或双烯加成反应生成配合物6或7,中间体6或7各自发生还原消除反应得到三取代苯,同时再生配合物2. 6,6'-二取代甲基的电子效应使得吡啶环上的氮原子电子云密度增大,配位能力增强,因而影响第三个炔烃分子取代4中联吡啶配体上的一个配位N原子生成钌杂环戊二烯-炔基配合物5.
三取代基苯有连三苯、偏三苯和均三苯三种同分异构体. Ru(Ⅲ)配合物催化末端炔烃发生环三聚反应时的反应机理中没有连三苯生成,而且生成偏三苯比生成均三苯的机会大,偏三苯应是主要的环三聚产物. 实验结果也只有偏三苯与均三苯两种同分异构体,而且生成的偏三苯多.如图3所示:末端炔烃是不对称的炔烃,在发生氧化偶联反应生成Ru杂环戊二烯配合物4时,由于取代基的位置不一样,会生成2,5-二取代Ru杂环戊二烯配合物9、3,4-二取代基Ru杂环戊二烯配合物10和2,4-二取代基Ru杂环戊二烯配合物11三种同分异构体.9经插入反应得到3,4,7-三取代基Ru杂环庚三烯配合物12和2,4,7-三取代基Ru杂环庚三烯配合物13;10经插入反应得到12和3,4,6-三取代的14;11得到13、14和2,4,6-三取代的15.经双烯加成反应,9转化为1,2,4-三取代基Ru杂降冰片二烯配合物16;10转化为2,3,5-三取代的17;11转化为1,3,5-三取代的18和1,2,5-三取代的19.中间体12、13、14、16、17、19发生还原消除反应生成8时,得到的是偏三苯;只有中间体15和18发生还原消除反应得到均三苯. 6,6'-二取代甲基的空间效应使得偏三苯的更易生成.
在催化环三聚反应中,加入乙腈或dmbp,均不利于环三聚产物的生成:加入乙腈,不利于炔烃取代配合物2中的水分子配体生成π-炔基配合物3;加入dmbp,炔烃分子难以取代配合物4中联吡啶配体上的配位N原子生成5.

图2 配合物2催化炔烃三聚形成苯的衍生物反应机理Fig.2 Mechanism of benzene derivatives formation from alkynes catalyzed by complex 2注: R=CH3(CH2)3,pH,C2H5OC(O).
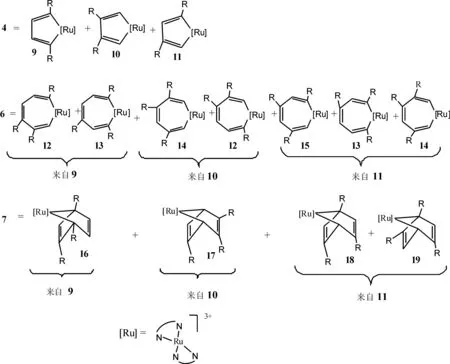
图3 催化环三聚产物的区域选择性Fig.3 Regioselectivity of catalyzed cylotrimerization
3 结 语
通过6,6'-二甲基-2,2'-联吡啶与RuCl3·3H2O的反应合成了三价钌配合物,进一步用银盐脱氯得到含联吡啶配体和二个水分子配体的钌配合物. 在催化末端炔烃的环三聚反应时,该钌配合物与炔烃反应依次生成中间体-炔基配合物、钌杂环戊二烯配合物和钌杂环戊二烯-炔基配合物,最后经过插入反应或双烯加成反应、还原消除反应得到取代苯,并再生含水配合物,完成催化循环. 6,6'-二甲基取代基的电子效应和空间效应的反应产率和区域选择性有较大的影响. 本实验对末端炔烃的催化环三聚机理研究,为设计炔烃的环三聚催化体系提供了理论依据.
致谢
感谢湖北省科技厅、湖北省教育厅和绿色化工过程教育部重点实验室的资助!
参考文献:
[1] Bianchini C, Masi D, Meli A, Peruzzini M, et al. Coupling of two Ethyne Molecules at Rhodium versus Coupling of two Rhodium Atoms at Ethyne[J]. Organometallics, 1991, 10: 636-645.
[2] Amer I, Schumann H, Ravindar V, et al. A Conv-enient Route to 1,3,5-Trisubstituted Benzenes via Rhodium Catalyzed Polymerization of Arylacetylenes[J]. J Mol Catal, 1993, 85: 163-171.
[3] Takeda A, Ohno A, Kadota I, et al. Regioselective Synthesis of 1,3,5-Unsymmetrically Substituted Benzenes via the Palladium-Catalyzed Cyclotrimeri-zation of 1,3-Diynes[J]. J Am Chem Soc, 1997, 119: 4547-4548.
[4] Liu Y B, Yan X Y, Yang N F, et al. Highly Regi-oselective Cyclotrimerization of Terminal Alkynes Catalyzed by Fe(II)Complexes Bearing 2-(Benzimidazolyl)-6-(1-(arylimino)ethyl) pyridines[J]. Catal Commun, 2011, 12: 489-492.
[5] Baxter R J, Knox G R, Moir J H, et al. Formation of Arenes and of Tetracarbonyl(hexatrienediyl)dicobalt (“Flyover”) Complexes from Co2(CO)8[J]. Organometallics, 1999, 18: 206-214.
[6] Vollhardt K P C. Transition-metal-catalyzed Acety-lene Cyclizations in Organic Synthesis[J]. Acc Chem Res, 1977, 10: 1-8.
[7] Mori N, Ikeda S.-I, Sato Y. Selective Cyclotrimeri-zation of Enones and Alkynes by a Nickel and Aluminum Catalytic System[J]. J Am Chem Soc, 1999, 121: 2722-2727.
[8] Takahashi T, Xi Z, Yamazaki A, et al. Cycloaddi-tion Reaction of Zirconacyclopentadienes to Alkynes: Highly Selective Formation of Benzene Derivatives from Three Different Alkynes[J]. J Am Chem Soc, 1998, 120: 1672-1680.
[9] Takahashi T, Kotora M, Xi Z J. Cycloaddition of Zirconacyclopentadienes to Alkynes using Copper Salts: Formation of Benzene Derivatives[J]. Chem Soc Chem Commun, 1995, 3: 361-362.
[10] Hara R, Guo Q X, Takahashi T. Reduction of Molybdenum(V) Chloride with Various Reducing Metals: Reactivity Correlations with the Desce-ndant Lewis Acids[J]. Chem Lett, 2000, 29: 140-141.
[11] Ardizzoia G A, Brenna S, LaMonica G, et al. Alkyne Oligomerization Catalyzed by Molybdenum(0) Complexes[J]. J Organomet Chem, 2002, 649: 173-180.
[12] Kaneta N, Hirai T, Mori M. Reaction of Alkyne Having Hydroxyphenyl Group with Mo(CO)6[J]. Chem Lett, 1995, 24: 627-628.
[13] 刘宇宙, 周立山, 席婵娟. Mo(CO)6催化炔烃三聚制备苯衍生物[J]. 化学学报, 2006, 64: 266-268.
LIU Yu-zhou, ZHUO Li-shan, XI Chan-juan. Mo(CO)6Catalyzed Cyclotrimerization of Alkynes: Formation of Benzene Derivatives[J]. Acta Chim Sinica 2006, 64: 266-268. (in Chinese)
[14] öztürk Bö. A Practical Ruthenium Based Catalytic System Bearing a Switchable Selectivity between the Dimerization and Cyclotrimerization Reactions of Alkynes[J]. Appl Catal A-Gen, 2012, 433-434: 214-222.
[15] Khedkar P, Kotha S. A Diversity-Oriented Appro-ach to Diphenylalkanes by Strategic Utilization of [2+2+2] Cyclotrimerization, Cross-Enyne Metathesis and Diels-Alder Reaction[J]. Eur J Org Chem, 2009, 5: 730-738.
[16] Farnetti E, Filipuzzi S. Directing Iridium-catalyzed C-C Bond Formation by Selection of the Ancillary Ligands: Polymerization and Cyclotrimerization of Alkynes[J]. Inorg Chim Acta, 2010, 363: 467-463.
[17] 尹传奇, 刘珺, 柏正武. 一种水溶性氢氧根钌配合物的合成及其对乙腈的催化水化作用[J]. 化学学报, 2011, 69: 2021-2025.
YIN Chuan-qi, LIU Jun, BAI Zheng-wu. Synthesis of a Water-Soluble Ruthenium Hydroxide Complex and Its Role in Catalytic Hydration of Acetonitrile[J]. Acta Chim Sinica, 2011, 69: 2021-2025. (in Chinese)
[18] Wilson S R, Wu Y H. A Study of Nickel-Catalyzed Coupling Reactions by Electrospray Ionization Mass Spectrometry[J]. Organometallics, 1993, 12: 1478-1480.
[19] Newkome G R, Pantaleo D C, Puckeet W E, et al. Complexes of Pd(II), Pt(II), Cu(II), Co(II) and Zn(II) Chloride with 6,6'-Dimethyl-2,2'-dipyridyl[J]. J inorg, nucl Chem, 1981, 43: 1529-1531.
[20] Ulucam G, Beynek N, Seller Z, et al. Synthesis, Characterization of Some Transition-Metal Comp-lexes of a New Heptadentate N5S2Schiff-Base Ligand and the Effects of These Metal Complexes on U2OS Cells Cytotoxicity and DNA Cleavage Activity[J]. Phosphorus, Sulfur, and Silicon, 2008, 183: 2237-2247.
[21] Parks J E, Wagner B E, Holm R H. Syntheses Employing Pyridyllithium Reagents : New Routes to 2,6- Disubstituted Pyridines and 6,6'-Disub-stituted 2,2'-Bipyridyls[J]. J Org Chem, 1973, 56: 53-56.
[22] Eggleston D S, Goldsby K A, Hodgson D J, et al. Structural variations Induced by changes in Oxid-ation State and their Role in Electron Transfer. Crystal and Molecular Structures of Cis-[Ru(bpy)2Cl2]·3.5H2O and cis-[Ru(bpy)2Cl2]Cl·2H2O[J]. Inorg Chem, 1985, 24: 4573.
[23] Durham B, Wilson S R, Hodgson D J, et al. Cis-Trans Photoisomerization in Ru(bpy)2(OH2)22+Crystal Structure of trans-[Ru(bpy)2(H2O)(OH)](ClO4)2[J]. J Am Chem Soc, 1980, 102: 600-607.
[24] Geetharani K, Tussupbayev S, Borowka J, et al. A Mechanistic Study of the Utilization of arachno-Diruthenaborane[(Cp*RuCO)2B2H6] as an Active Alkyne-Cyclotrimerization Catalyst[J]. Chem Eur J, 2012, 18: 8482-8489.
猜你喜欢
上海师范大学学报·自然科学版(2023年1期)2023-06-30 07:18:19
石油炼制与化工(2017年1期)2017-04-06 07:18:53
赣南师范大学学报(2016年3期)2016-07-18 05:51:05
中国塑料(2015年9期)2015-10-14 01:12:21
应用化工(2014年1期)2014-08-16 13:34:08
石油炼制与化工(2014年4期)2014-04-06 22:11:17
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:06
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年12期)2014-02-28 17:33:51
无机化学学报(2014年10期)2014-02-28 17:33:16
