
用于碳烟燃烧的Cu0.05Ce0.95O催化剂活性氧物种的研究——等离子体的强化效应
2013-01-18 07:00欧阳杰宏林俊敏刘有发付名利黄碧纯吴军良陈礼敏叶代启
中国环境科学 2013年2期
欧阳杰宏,林俊敏,刘有发,付名利,2,黄碧纯,吴军良,陈礼敏,叶代启,2,3,4*
(1.华南理工大学环境科学与工程学院,广东 广州 510006;2.华南理工大学工业聚集区污染控制与生态修复教育部重点实验室,广东 广州 510006;3.华南理工大学广东省大气环境与污染控制重点实验室,广东 广州 510006;4.华南理工大学广东高校大气污染控制工程研究中心,广东 广州 510006)
颗粒物(PM)是柴油机排放的主要污染物之一,其排放量可达到汽油机排放的 50~80倍[1].碳烟是颗粒物的主要成分,其着火温度高达 550~650℃,而典型的柴油机排气温度很低,约在 200~400℃[2-3],在该温度区间内碳烟很难实现高效燃烧,起燃活性很低.由于CeO2具备优良的氧储存能力(OSC),铈基催化剂被广泛报导和研究.近年来,介质阻挡放电(DBD)方式能在较宽的电压范围内产生高能量的等离子体,广泛应用于材料改性[4]、降解有机物[5-6]、选择性催化还原 SCR[7]等方面,相对而言介质阻挡放电-催化相结合技术应用于碳烟氧化燃烧的研究,尤其是等离子体对催化剂活性氧物种的研究还不是非常深入.
故本文采用前期工作[8]优选的络合燃烧法制备的 Cu0.05Ce0.95-CA催化剂和一段式介质阻挡放电等离子体-催化反应器.运用程序升温氧化(TPO)反应比较不同电压负载下碳烟催化燃烧的活性.利用程序升温脱附(O2-TPD)、程序升温还原(H2-TPR)、X射线衍射(XRD)、比表面积分析(BET)和 X射线光电子能谱仪(XPS)等进行表征,重点探讨了等离子体对碳烟燃烧的铜铈复合氧化物催化剂活性氧物种的强化效应.
1 实验方法
1.1 催化剂制备
采用柠檬酸络合燃烧法制备复合氧化物催化 剂 Cu0.05Ce0.95-CA[8].以 Ce(NO3)3·6H2O 和Cu(NO3)2·3H2O 作为前驱物,按 Ce:Cu 摩尔比为19:1配比,于70℃添加柠檬酸使pH值调为1后,升温至 100℃.混合物形成胶状粘稠物后,快速升温至150℃,直至燃烧完毕取出,碾磨至粉末,然后置于马弗炉中于450℃焙烧5h制得.
1.2 活性评价
图1为介质阻挡放电-催化一体式反应活性评价装置.等离子体-催化一段式反应器由等离子发生系统和催化反应管式炉组成.石英管内、外径分别为 12mm、16mm,长度为 400mm,石英管插入一根直径为2mm的不锈钢电极作为电极,管外壁缠绕不锈钢丝作为地极.两级间可通入高压电,通过介质阻挡方式放电产生等离子体,放电参数由电源自带数显表可得.催化剂与模拟碳烟(Printex®U,德国 Degussa公司生产,平均原生粒径为25nm)按质量比为9:1的比例混合,接触方式为松散接触,总质量为 1000mg,在坩埚中搅拌混合后,装入石英反应管中,催化剂碳烟混合物两端装填石英棉固定.所选用的反应模拟气体为O2(10%)和 Ar(90%,平衡气),分别由质量流量计控制,气体于缓冲瓶中混合后由转子流量控制,恒定于 100mL/min,空速为 100mL/(min·g).
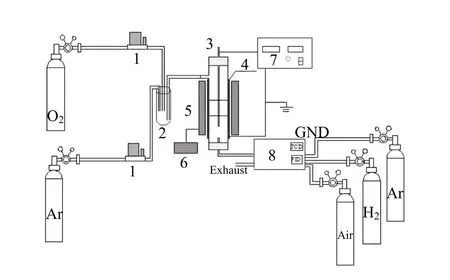
图1 介质阻挡放电-催化一体式反应活性评价装置Fig.1 DBD-catalysis one-piece TPO equipment
通入纯Ar气在300℃吹扫1h,降至100℃后切换到反应气并在反应管两级间通入一定电压,进行程序升温,从 100℃升到 800℃,升温速率4℃/min.反应出口气体 CO2和 CO每隔 7min由上海科创 GC-900A型气相色谱检测,检测 CO2和CO利用Ni转化炉将CO2和CO转化成CH4,色谱担体为TDX-01,检测器为FID,CO2和CO的检出限为5×10-6.
以碳烟的燃烧温度,CO2的选择性和理论碳脱除率来评价反应活性.Ti为反应尾气 CO2浓度达到5000×10-6的温度[16];Tm为CO2浓度达到最大值时的峰值温度;Tc为碳烟燃尽的温度.Ti越低,表示催化剂有着更好的起燃活性.△T=Tm-Ti反映了催化剂不同条件下碳颗粒燃烧速度的差异,△T越小,说明反应的整体燃烧速率越高.Sco2为温度达到Tm时CO2的选择性.ηc通过出口CO2浓度与检测时间段乘积积分与实际投入到体系的碳烟量(100mg)的比,即理论的碳脱除率.
1.3 催化剂表征
程序升温脱附(O2-TPD):预处理,催化剂分别在放电和无放电情况进行氧气吸附至饱和,放电电压为9kv.采用美国麦克Auto-Chem II 2920化学吸附仪.催化剂用量为 100mg.首先 He气(50mL/min)在 400℃吹扫 60min,自然降至 50℃,再用5%O2/He(50mL/min)吸附60min,切换成He气(50mL/min),待基线稳定后从 50℃升温至800℃进行程序脱附实验,检测器为TCD.
程序升温还原(H2-TPR):采用美国麦克Auto-Chem II 2920化学吸附仪.催化剂用量为100mg. AR 气(50mL/min)在 400℃吹扫 60min.自然降至 50℃.切换成 10%H2/AR(50mL/min)待基线稳定后从 50℃升温至 800℃进行程序还原实验,检测器为TCD.
X射线衍射(XRD):采用德国Bruker公司D8 ADVANCEX射线衍射仪.实验条件:铜靶,入射线波长 0.15418nm,Ni滤波片,管压 40KV,管流40mA,扫描步长 0.02°,扫描速度 0.1s/步,狭缝DS0.5°RS8mm(对应 LynxExe阵列探测器),扫描范围(2θ)为 20°~80°.
比表面积分析(BET):采用美国麦克 ASAP-2020M 全自动比表面积分析仪.样品使用量为100~300mg.经 300℃抽真空预处理 120min,以 N2为吸附质,在-196℃进行测定.
X 射线光电子能谱仪(XPS):采用 VG Multilab 2000测定,其测试条件为 Mg Kα射线(hν=1253.6 eV),C 1s校准结合能284.6eV.
2 结果与讨论
2.1 活性评价
图2为不同条件下碳烟燃烧TPO反应检测的CO和CO2的出口浓度,表1为Ti,Tm,Tc和△T结果.可以观察到无论等离子体是否存在,碳烟燃烧的起燃温度和峰值温度变化不大(Ti,nopl=364.1℃,Ti,pl= 349.5℃与Tm,nopl= 517.2℃,Tm,pl=516.8℃),说明本实验条件下等离子体在无催化剂的情况下很难提高碳烟燃烧的活性.等离子体存在时,TPO反应在 220℃已经观察到碳烟转化物,而无等离子体存在的TPO反应在297℃才能观察到,推测是O2在放电区形成的高氧化物种O自由基甚至O3可在低温段氧化碳烟,然而此氧化过程主要是不完全氧化,生成的是 CO,降低了CO2的选择性(Sco2,nopl=38.95%,Sco2,pl=30.44%).
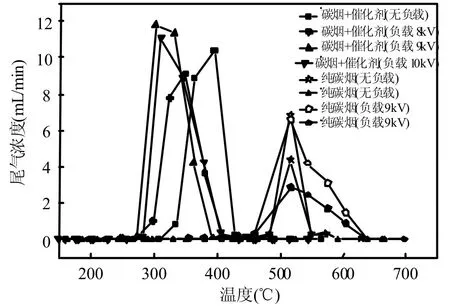
图2 不同条件下碳烟燃烧TPO曲线Fig.2 TPO curves of soot oxidation under different conditions

表1 不同条件下碳烟燃烧的Ti ,Tm , Tc和△TTable 1 Ti ,Tm , Tc and △T for soot combustion under different conditions
Cu0.05Ce0.95O-CA催化剂引入到碳烟燃烧体系后,起燃温度Ti和峰值温度Tm降低到308.2℃和395.5℃,燃烧区间△T下降为87.3℃,燃烧速率加快,大大促进碳烟燃烧的活性.同时将等离子体引入到碳烟催化燃烧体系后,Ti,Tm和△T进一步降低.在负载电压为 9kv的条件时效果最好:Ti=243.1℃,Tm=302.8℃,△T=59.7℃,理论的碳脱除率为95.12%,起燃温度已经十分接近柴油机排气温度区间(200~400℃)的下限,并表现出良好的碳烟催化活性.与无催化剂引入体系的情况相似,等离子体存在下在低温区约 180℃已经有少量的碳烟转化物,且低温氧化为完全氧化反应,无 CO生成,有研究[22]指出,原因可能产生了高活性的C*[O] .本实验条件引入催化剂后,TPO反应几乎不产生CO,使得CO2选择性接近100%,说明络合燃烧法制备的Cu0.05Ce0.95O-CA是一种理想的、高选择性的催化剂,也同样说明了碳烟燃烧过程的选择性主要由催化剂[24]决定.
2.1 H2-TPR和O2-TPD
图3为利用不同方法预处理的催化剂的H2-TPR图.表2为H2-TPR实验结果.图3中a峰归结为表面高度分散的CuO的还原峰,b峰为进入CeO2晶格的Cu2+的还原峰[33-34].经过等离子体处理后,催化剂表面可还原物种种类并没有产生变化,显著的变化表现在还原温度降低和还原峰耗氢量增加.与无等离子体处理相比,等离子体(电压9kV)预处理过的催化剂2个还原峰温度分别降低了 14.2℃和 9.5℃,耗氢量分别提高了29.67%和 48.48%.由此可得,等离子体可以提高催化剂表面游离态的CuO的分散度[32],并使得更多的 Cu2+进入 CeO2晶格[35],形成更多的具有丰富氧空位的Cuy2+Ce1-y4+O2-y2-□y固熔体.
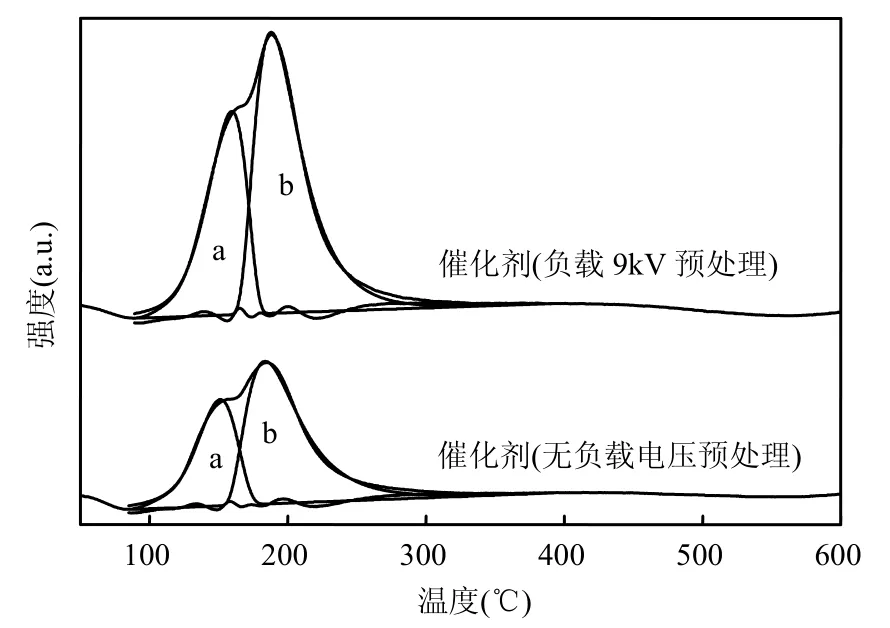
图3 Cu0.05Ce0.95-CA催化剂H2-TPRFig.3 H2-TPR of Cu0.05Ce0.95-CA catalyst
过渡金属催化剂表面存在 3种氧物种为α氧、β氧和γ氧.其中α氧(脱附温度<350℃)属于普通分子吸附氧O2-;β氧(脱附温度 350~750℃)属于与氧缺位有关的化学吸附氧 O-;γ氧(脱附温度>750℃)属于晶格氧O2-,是催化剂表面结构中的分子氧 O2-[13-14].研究[8]发现,掺杂少量铜的铈基氧化物催化剂比单一的氧化铈催化剂有着更强的氧脱附能力,原因是 Cu2+部分取代Ce4+,导致 CeO2的晶格缺陷,产生更多的表面氧空穴,强化了活性氧在催化剂表面的积累[41],同时也促进了Ce4+↔Ce3+的振荡[16,40],最终促进了碳烟的燃烧.
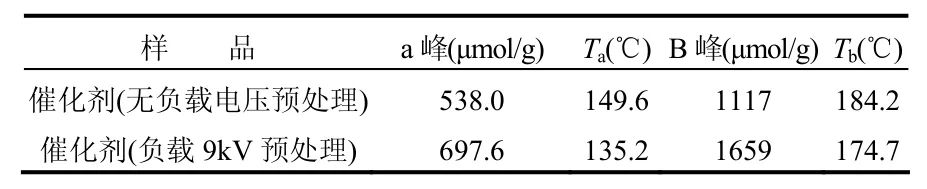
表2 H2-TPR实验中Cu0.05Ce0.95-CA催化剂耗氢量Table 2 Hydrogen consumption of Cu0.05Ce0.95-CA catalysts by H2-TPR
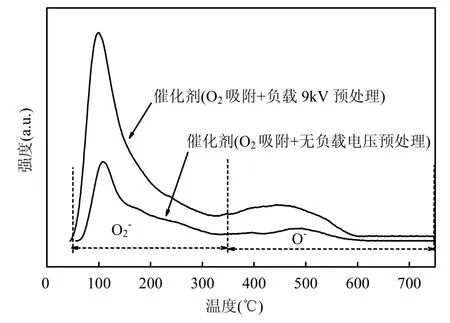
图4 Cu0.05Ce0.95-CA催化剂O2-TPDFig.4 O2-TPD of Cu0.05Ce0.95-CA catalysts
图4为Cu0.05Ce0.95-CA催化剂O2-TPD图.经过等离子体预处理的催化剂表现出更加优异的氧脱附性能.无论是分子吸附氧 O2-还是化学吸附氧O-,经过等离子体预处理后的氧脱附量远高于无等离子体预处理时的情况.表 3为仪器自带软件Grams/32计算的不同预处理方法催化剂的分子吸附氧 O2-和化学吸附氧 O-的脱附量,单位为μmol/g.在等离子作用下,催化剂储氧容量增大,在催化剂表面可能生成更多的活性氧物种[36],其流动、迁移和转化促进催化剂产生更多的氧空穴[37-39],如此循环,持续促进碳烟催化燃烧.2种情况的脱附峰温度相近可以看出,等离子体只是增加了活性物种的数目[16],与H2-TPR结果一致.

表3 O2-TPD实验中Cu0.05Ce0.95-CA催化剂分子吸附氧O2- 和化学吸附氧O-的脱附量Table 3 Oxygen desorption capacity of Cu0.05Ce0.95-CA catalysts by O2-TPD
2.2 BET和XRD
图5为不同预处理条件的Cu0.05Ce0.95-CA催化剂的XRD谱图,表4为催化剂的BET和XRD结果.由 XRD 图可以观察到,各个样品均呈现出CeO2的立方相萤石结构,说明等离子体并不会改变催化剂的整体晶体结构[21],与 H2-TPR结果一致.新鲜 Cu0.05Ce0.95-CA催化剂平均晶粒尺寸为 14.5nm,属于纳米级催化剂.比较明显的几个特征峰,约在 2θ=28.5°,33.1°,47.6°,56.5°是 CeO2的特征峰,对应的是立方相萤石结构的 111,200,220,311 晶面[9];此外在 59.2°,69.5°,76.8°和 79.1°附近出现的微小特征衍射峰对应的是 CeO2的222,400,331和 420晶面结构[10,15],与标准谱图JCPDS# 78-694相吻合.Cu0.05Ce0.95-CA催化剂均没有观察到CuO的晶相衍射峰(CuO的特征峰2θ约在 35.5°,38.7°,48.7°,61.5°),原因可能存在三点:铜的添加量太少,在前期研究[8,18]中发现,当Cu的摩尔比例达到75%以上时才能出现明显的CuO的晶相衍射峰;与铈结合形成复合氧化物[17];CuO在CeO2表面的高度分散所致[11-12].

表4 Cu0.05Ce0.95-CA 催化剂的BET和XRD结果Table 4 BET and XRD results of Cu0.05Ce0.95-CA catalysts
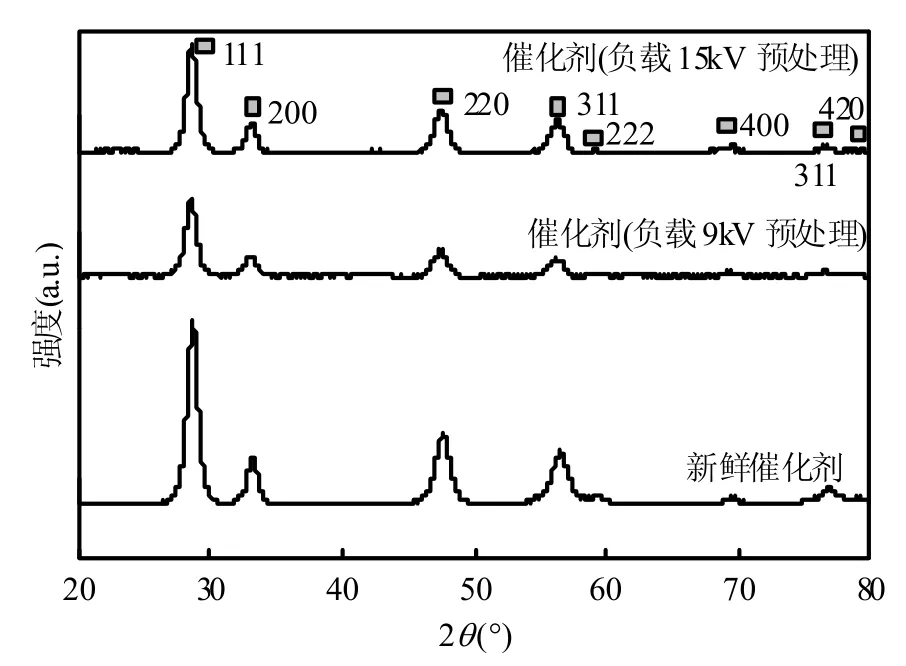
图5 Cu0.05Ce0.95-CA 催化剂的XRD谱Fig.5 XRD Patterns of Cu0.05Ce0.95-CA Catalysts
经过等离子体预处理后,衍射峰宽化现象十分明显,晶体的结晶度大大降低,催化剂平均晶粒尺寸进一步减少到 12.50nm(负载 9kV 电压)和11.20nm(负载15kV电压),其CeO2的111晶面衍射角进一步向低角度位移(28.64°→28.40°).出现这种现象的原因说明在等离子体预处理后,Cu2+(离子半径 0.072nm)进一步进入 CeO2晶胞,掺杂形成具有丰富氧空位的Cuy2+Ce1-y4+O2-y2-□y固熔体[17].CeO2晶胞的膨胀诱导了结构缺陷,产生更多氧空穴,提高了催化剂的非晶格氧(分子吸附氧O2-和化学吸附氧 O-)的流动性[23],与前文的O2-TPD结果相吻合.BET结果显示经等离子体处理后,比表面积由63.75m2/g提高到82.89m2/g和 80.04m2/g,孔容由 0.1478mL/g提高到0.2224mL/g和0.2188mL/g.更大的比表面积和孔容意味着催化剂表面能够提供足够的活性位[32],促进催化剂氧化碳烟燃烧的催化活性,与H2-TPR 结果相符合.有研究[37]指出,更大的比表面积意味着催化剂有着更强的储氧能力(OSC),与O2-TPD结果一致.
2.3 XPS
表5为不同条件TPO处理前后的表面原子的XPS结果.TPO反应前后表面原子含量对比可以发现,C原子含量有所降低,等离子体存在时下降幅度更大,与TPO反应碳脱除率随等离子体引入提高的现象相吻合,等离子体可以有效减缓催化剂表面碳烟残留现象.Ce,Cu和O原子在TPO反应后均有不同程度的提高,不仅仅表现在原子比例(%)的提高,也表现在强度(CPS)的提高.等离子体在催化剂表面的作用主要表现在两方面:一是等离子体作用下高氧化活性物种(氧自由基O·和臭氧 O3等)吸附在催化剂表面,直接参与催化反应[25];二是等离子高能电子的作用下,能在不改变催化剂整体的晶体结构情况下,改变了催化剂表面的活性物种的组成[26]:

表5 TPO反应前后混合物表面原子的百分比(%)Table 5 Surface species percentage of mixture before and after TPO by XPS(%)
等离子体使高氧化性物种(O·和 O3等)吸附在Cu0.05Ce0.95-CA催化剂表面(氧空位),直接参与碳烟燃烧反应,使催化剂具备了低温氧化活性,与 TPO 结果相吻合;此外,等离子体可作用于 Cu0.05Ce0.95-CA催化剂表面进行改性,Cu2+充分进入到 CeO2的晶胞,产生更多的Cuy2+Ce1-y4+O2-y2-□y固溶体,提高了氧物种在催化剂表面的流动性,增加了活性中心的数目,与O2-TPD和XRD结果吻合.
图6为TPO处理前后的Ce 3d和O 1s的XPS谱图.对XPS谱图进行分峰拟合处理,Ce,O的各物种成分含量的百分比列于表 6中.Ce 3d XPS谱图中u和v轨道分别代表Ce的3d3/2和3d5/2轨道.v′, u′轨道代表的是 Ce3+;v′′、v′、v、u′′、u′、u 轨道代表的是 Ce4+[27].O 1s谱图中528.8±0.5eV 为晶格氧 O2-,530.7±0.5eV 为与原子吸附氧O-,532±0.5eV为分子吸附氧O-[28].

图6 Cu0.05Ce0.95-CA不同电压TPO反应后的XPS谱Fig.6 XPS spectra for Cu0.05Ce0.95-CA catalysts after TPO with different voltage
在TPO反应前后Ce4+/Ce3+比例存在不同程度的提高,说明表面部分Ce4+还原成Ce3+,使Ce3+阳离子周围形成缺陷,由氧空穴进行补偿[29-30].等离子体存在下TPO反应Ce4+/Ce3+比例明显降低,说明在等离子体存在下催化剂表面 Ce3+正离子增多,产生更多的氧空穴,提高了催化剂的氧的流动性,与XRD和O2-TPD结果相符.
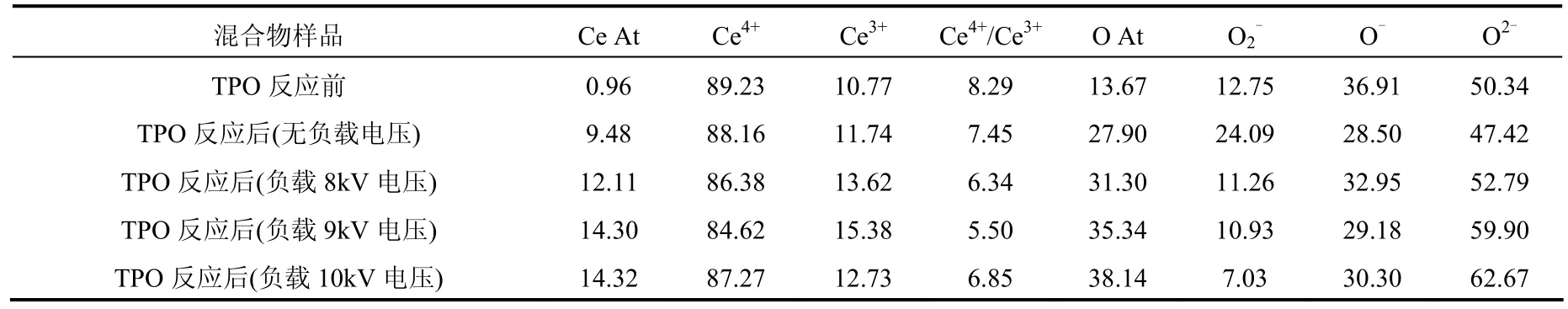
表6 TPO前后混合物的表面金属铈物种和氧物种的百分比(%)Table 6 Surface Oxygen and Cerium species percentage of mixture before and after TPO by XPS(%)
表面的 O物种结果显示,在等离子体存在下,TPO反应前后吸附氧(分子吸附氧 O2-和化学吸附氧 O-)下降,晶格氧 O2-含量在提高.结合 Ce物种的含量变化、XRD和O2-TPD结果说明在等离子体存在下,主要供氧体可能是 Ce4+/Ce3+氧化还原振荡产生在 Ce3+周围补偿型氧空位[29-31]以及放电区内形成并吸附在催化剂表面的高氧化性物种(O·和O3等)[22,25-26].
在等离子作用下,使得催化剂表面游离的Cu2+不断进入 CeO2的晶胞,形成更多的Cuy2+Ce1-y4+O2-y2-□y固溶体,促进了 Ce4+/Ce3+氧化还原振荡,产生更多的补偿氧空穴,同时放电区O2形成的高氧化物种(O·和 O3等)吸附在催化剂表面氧空穴形成活性氧,与碳烟颗粒发生反应消耗(低温生成高活性的C*[O],促进低温氧化反应),氧空穴的活性氧由高氧化物种补充,不断循环,保持反应进行.
3 结论
3.1 在等离子体放电下, Cu0.05Ce0.95-CA催化剂氧化碳烟的活性得到显著提高.电压为 9kV时,碳烟的起燃温度由 308.2℃下降到 243.1℃,燃烧峰值温度由 395.5℃下降至 302.8℃,燃烧速率提升,碳脱除率计算值提升至95.12%.
3.2 在等离子体放电下, Cu0.05Ce0.95-CA催化剂仍能保持 CeO2的立方萤石结构;Cu0.05Ce0.95-CA催化剂有着更小的晶粒粒径和更大的比表面积、孔容;其提高催化剂表面游离态的CuO的分散度,并使得更多的 Cu2+进入 CeO2晶格,形成更多的
Cuy2+Ce1-y4+O2-y2-□y固熔体.
3.3 等离子体作用下催化剂储氧容量增大,在催化剂表面生成更多的活性氧物种,其流动、迁移和转化促进催化剂产生更多的氧空穴,如此循环,持续促进碳烟催化燃烧.
[1] 吴银山,林 寅,林 枫.柴油车与汽油车的排气污染和其它性能比较 [J]. 环境, 2008,(S1):41-44.
[2] Krishna K, Bueno-LÓpez A, Makkee M, et al. Potential rare earth modified CeO2catalysts for soot oxidation I:Characterisation and catalytic activity with O2[J]. Applied Catalysis B:Environmental,2007,(75):189-200
[3] 常仕英,吴庆伟,杨冬霞,等.柴油车碳烟的燃烧特性及动力学研究 [J]. 内燃机学报, 2009,27(3):255-258.
[4] 唐晓亮,冯贤平,黎志光,等.常压介质阻挡放电等离子体发射光谱诊断及其在材料表面改性中的应用 [J]. 光谱学与光谱分析,2004,24(11):1437-1440.
[5] Huang H B, Ye D Q, Leung D Y C. Abatement of toluene in the plasma-driven catalysis: Mechanism and reaction kinetics [J].IEEE Transactions on Plasma Science, 2011,39(3):877-882.
[6] 史恒超,王文春,杨德正,等.介质阻挡放电中 OH 自由基对甲醛脱除的影响 [J]. 物理化学学报, 2011,27(8):1979-1984.
[8] Fu Mingli, Yue Xianghui, Ye Daiqi, et al.Soot oxidation via CuO doped CeO2catalysts prepared using coprecipitation and citrate acid complex-combustion synthesis [J]. Catalysis Today, 2010,153(3):125-132
[9] Cousin R, Capelle S, Abi-Aad E, et al.Copper-vanadium-cerium oxide catalysts for carbon black oxidation [J]. Catalysis B:Environmental, 2007,70(1):247-253.
[10] Larachi F, Pierre J, Adnot A, et al.Ce 3d XPS study of composite CexMn1-xO2-ywet oxidation catalysts [J]. Applied Surface Science,2002,195(1):236-250.
[11] George Avgouropoulos, Theophilos Ioannides.Effect of synthesis parameters on catalytic properties of CuO-CeO2[J]. Catalysis B:Environmental, 2006,67(1):1-11.
[12] Hocevar S, Opara Krašovec U, Orel B, et al. CWO of phenol on two differently prepared CuO-CeO2 catalysts [J]. Applied catalysis B: Environmental, 2000,28(2):113-125.
[13] Zhu Junjiang, Zhao Zhen, Xiao Dehai, et al.Study of La2-xSrxCuO4(x= 0.0, 0.5, 1.0) catalysts for NO+CO reaction from the measurements of O2-TPD, H2-TPR and cyclic voltammetry [J]. Journal of Molecular Catalysis A: Chemical,2005 238(1):35-40
[14] Jiang Xiaoyuan, Zhou Renxian, Pan Ping,et al.Effect of the addition of La203on TPR and TPD of CuO/ɤ-Al2O3catalysts [J].Applied Catalysis A: General, 1997,150(1):131-141.
[15] 韦岳长,刘 坚,赵 震,等. Co0.2/Ce1-xZrxO2催化剂的制备、表征及其催化碳烟燃烧反应性能 [J]. 催化学报, 2010,31(3):283-288.
[18] 陈 瑜.二氧化硫气氛下氧化物催化剂去除柴油机排气碳烟的研究 [D]. 广东:华南理工大学学位论文, 2008.
[19] 何 迈,方 萍,罗孟飞. CuO/CeO2-Al2O3催化剂中CuO高温迁移和固相反应的研究 [J]. 中国稀土学报, 2005,23(6):674-678
[20] 晏冬霞,王 华,李孔斋,等.Ce1-xFexO2复合氧化物的结构及其催化碳烟低温燃烧性能 [J]. 物理化学学报, 2010,26(2):331-337.
[21] 朱艳茹.低温等离子对镍基催化剂的改性研究 [D]. 天津:天津大学硕士学位论文, 2005.
[22] 裴梅香,林 赫,上官文峰,等.等离子体辅助同时催化去除柴油机 NOx和碳烟的试验研究 [J]. 工程热物理学报, 2005,26(5):879-882.
[23] 晏冬霞,王 华,李孔斋等.铈铁锆三元复合氧化物上碳烟的催化燃烧 [J]. 燃烧化学学报, 2011,39(3):229-234.
[24] Zheng Rongyao, Cai Yixi, Wang Pan. Experiment on the removal of soot emissions from diesel engine with non-thermal plasma assisted catalyst technology [J]. 2011 International Conference on Electric Information and Control Engineering (ICEICE):5012-5015.
[25] Hueso J L, Cotrino J, Caballero A, et al.Plasma catalysis with perovskite-type catalysts for the removal of NO and CH4from combustion exhausts [J]. Journal of Catalysis, 2007,247(2):288-297.
[26] Ekaterina S Lokteva, Aleksey E Lazhko, Elena V Golubina , et al.Regeneration of Pd/TiO2catalyst deactivated in reductive CCl4transformations by the treatment with supercritical CO2,ozone in supercritical CO2or oxygen plasma [J]. The Journal of Supercritical Fluids, 2011,58(2):263-271.
[27] Ma Bin, Li Yao, Su Ke. Characterization of ceria-yttria stabilized zirconia plasma-sprayed coatings [J]. Applied Surface Science,2009,255(16):7234-7237
[28] Wei Yuechang, Liu Jian, Zhao Zhen, et al. The catalysts of three-dimensionally ordered macroporous Ce1-xZrxO2-supported gold nanoparticles for soot combustion:The metal-support interaction [J]. Journal of Catalysis, 2012,287:13-29
[29] Martín S Gross, María A Ulla, Carlos A Querini. Diesel particulate matter combustion with CeO2as catalyst. Part I:System characterization and reaction mechanism [J]. Journal of Molecular Catalysis A:Chemical, 2012, 352:86-94.
[30] Mogens Mogensen, Nigel M Sammes, Geoff A Tompsett.Physical,chemical and electrochemical properties of pure and doped ceria [J]. Solid State Ionics, 2000,129(1):63-94.
[31] Zhu Ling, Yu Junjie, Wang Xuezhong. Oxidation treatment of diesel soot particulate on CexZr1-xO2[J]. Journal of Hazardous Materials, 2007,140(1):205-210.
[32] Shi Limin, Chu Wei, Deng Siyu, et al.Catalytic properties of Cu/Co/Zn/Zr oxides prepared by various methods [J]. Journal of Natural Gas Chemistry,2008,17(4):397-402.
[33] Albin Pintar, Jurka Batista, Stanko Hocevar. TPR, TPO, and TPD examinations of Cu0.15Ce0.85O2-ymixed oxides prepared by co-precipitation, by the sol-gel peroxide route, and by citric acid-assisted synthesis [J]. Journal of Colloid and Interface Science, 2005,285(1):218-231.
[34] Mai Hailing, Zhang Dengsong, Shi Liyi, et al. Highly active Ce1-xCuxO2nanocomposite catalysts for the low temperature oxidation of CO [J]. Applied Surface Science, 2011,257(17):7551-7559.
[35] Hua Wei, Jin Lijun, He Xinfu, et al. Preparation of Ni/MgO catalyst for CO2reforming of methane by dielectric-barrier discharge plasma [J]. Catalysis Communications, 2010,11(11):968-972.
[36] Albin Pintar, Jurka Batista, Stanko Hocevar.Nanostructured CuxCe1-xO2-ymixed oxide catalysts:Characterization and WGS activity tests [J]. Journal of Colloid and Interface Science, 2007,307(1):145-157.
[37] Zhu Ling, Yu Junjie, Wang Xuezhong. Oxidation treatment of diesel soot particulate on CexZr1-xO2[J]. Journal of Hazardous Materials, 2007,140(1):205-210.
[38] Lin He, Huang Zhen, Shangguan Wenfeng, et al.Temperatureprogrammed oxidation of diesel particulate matter in a hybrid catalysis-plasma reactor [J]. Proceedings of the Combustion Institute, 2007,31(2):3335-3342.
[39] Andrew S D’Souza, Shane Leiphart, Carlo G Pantano. RF-plasma treatments of surface-conductive alkali-lead silicate glass and microchannel plate devices [J]. Applied Surface Science, 1999,148(1):126-132.
[40] Wei Yuechang, Liu Jian, Zhao Zhen, et al. The catalysts of three-dimensionally ordered macroporous Ce1-xZrxO2-supported gold nanoparticles for soot combustion [J]. Journal of Catalysis,2012,287:13-29.
[41] Gloria Preda, Gianfranco Pacchioni.Formation of oxygen active species in Ag-modified CeO2catalyst for soot oxidation: A DFT study [J]. Catalysis Today, 2011,177(1):31-38.
猜你喜欢
温州大学学报(自然科学版)(2022年2期)2022-05-30
空间科学学报(2021年6期)2021-03-09
潍坊学院学报(2020年2期)2021-01-18
科学大众(中学)(2019年3期)2019-05-17
汽车观察(2018年10期)2018-11-06
北京航空航天大学学报(2017年7期)2017-11-24
北京航空航天大学学报(2017年7期)2017-11-24
北京航空航天大学学报(2017年2期)2017-11-24
制导与引信(2017年3期)2017-11-02
科技知识动漫(2017年1期)2017-02-06
