
叶酸在缺血/再灌注损伤中的作用
2013-01-11 06:45:14王若禺崔术楠邢宏义
山东第一医科大学(山东省医学科学院)学报 2013年7期
王若禺 崔术楠 邢宏义
(1.华中科技大学附属协和医院,武汉430022;2.华中科技大学附属协和医院神经内科,武汉 430022)
叶酸是B族维生素的一种,本身不具有生物活性,叶酸在肝脏转换成二氢叶酸再转变为四氢叶酸并具有生物活性[1]。同型半胱胺酸并不是从食物中获得的,而是甲硫氨酸经代谢得到的。缺乏维生素B6、叶酸和B12可导致高同型半胱胺酸血症。高同型半胱胺酸血症的患者发生血栓形成和心脑血管疾病的概率相比正常人群更高[2]。叶酸在缺血/再灌注的多个环节中起保护作用,该文将通过高同型半胱胺酸血症、核因子κB(nuclear factorκB,NF-κB)激活以及内皮一氧化氮合酶(endothelial nitric oxide synthase, eNOS)解联合等环节阐述叶酸缺乏可促进缺血/再灌注损伤。
1 叶酸缺乏导致缺血/再灌注损伤
1.1高同型半胱胺酸血症 目前的观点认为,高同型半胱胺酸血症是心血管疾病的重要危险因素之一,高同型半胱胺酸血症可导致血管功能异常。有研究指出,在体内实验中,同型半胱胺酸促进单核细胞驱化蛋白1(monocyte chemoattractant protain-1,MCP-1)、血管内皮细胞黏附分子1和E-选择素的表达,导致单核细胞黏附于血管内皮[3]。在实验中,实验组喂养普通饮食配合1.7%的甲硫氨酸饮食四周,导致血液中同型半胱胺酸浓度急剧升高,同时单核细胞的驱化和黏附运动相比对照组(无甲硫氨酸饮食)也明显增加(292% vs.100%)。免疫组织化学的结果表明高同型半胱胺酸的大鼠,其血管内皮MCP-1的含量明显增多,这表明同型半胱胺酸能够诱导MCP-1表达。在另一个实验中,实验中被预先注入MCP-1的抗体再用甲硫氨酸喂养,与对照组相比单核细胞的驱化运动无显著性差异[3]。用血管内皮细胞黏附分子1和E-选择素预先处理的实验中也得到了相似的结果[3-4]。以上这些结果表明,高同型半胱胺酸血症通过激活MCP-1、血管内皮细胞黏附分子1和E-选择素的表达来驱化单核细胞黏附于血管内皮,从而促进血管内皮的损伤。
1.2DNA损伤以及高能磷酸化合物耗尽 多个研究表明,大脑缺血/再灌注损伤可以导致缺血区域DNA的损失,但在心肺的缺血/再灌注损伤中,还未有DNA损伤的报道[5-6]。Endres等[4]发现,同型半胱胺酸可以加重脑组织的再灌注损伤。AP位点(Apurinic/apyrimidinic abasic sites, AP sites)是一种检测DNA损伤程度的试剂盒,实验组予以无叶酸饮食,对照组给予叶酸饮食,两组都制作大脑中动脉栓塞模型,再灌注后实验组AP sites的染色程度远远高于对照组,说明无叶酸饮食可以加重DNA的损伤。最主要的原因是叶酸的缺乏导致胸嘧啶合成的缺乏。叶酸是参与转移甲基的重要辅酶,而胸腺嘧啶合成需要转甲基过程的参与。叶酸的缺乏可导致胸腺嘌呤合成不足,那么尿嘧啶可能会被误编入DNA中,从而导致DNA的损伤。同样,黄嘌呤的合成也需要叶酸及转甲基的参与,黄嘌呤再经过加工可以合成腺苷酸(adenosine monophosphate,AMP)和鸟苷酸(guanosine monophosphate,GMP)。叶酸的缺乏不仅导致胸腺嘧啶的缺乏,还可以影响AMP和GMP的合成。AMP是三磷酸腺苷(adenosine triphosphate,ATP)的合成原料,GMP是环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)第二信使的合成原料,同时另一种第二信使环磷酸腺苷的合成也受到影响。叶酸的缺乏可以降低细胞内AMP和GMP的浓度,导致第二信使合成的量不足,从而影响细胞间通信的效率,但是还没有客观的指标可以证明这一观点。一氧化氮(nitric oxide,NO)是一种重要的扩血管药物,它通过促进血管平滑肌cGMP的合成最终促进血管的舒张[7]。叶酸的缺乏可同时导致NO和cGMP的减少,从而影响再灌注时血管的舒张,影响缺血区血流灌注。
在缺血/再灌注中受到损伤的细胞,需要用AMP、GMP和胸苷酸来修复受损的DNA;缺少这些原料修复过程会受阻,那么细胞将会开始凋亡的过程。在缺血/再灌注损伤中,凋亡是组织受损伤的一种重要的形式,Caspase蛋白家族是执行细胞凋亡的重要通路(图1)。如果DNA损伤超过一定的阈值,Caspase蛋白就会被激活,然后活化更多的蛋白酶,将DNA水解成200个碱基对或其倍数大小的片段,通过DNA凝胶电泳可以发现这一现象。Endres等发现,用了Caspase的抑制剂后,缺血/再灌注损伤动物(大脑中动脉栓塞模型)的脑组织损伤明显减轻[4]。由于叶酸的缺乏可以导致DNA损伤,而缺血/再灌注损伤是加重DNA损伤的重要过程,所以叶酸缺乏的动物模型缺血/再灌注损伤的程度更重(图2)。
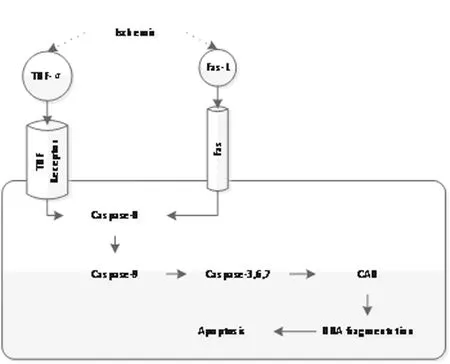
缺血的过程使得TNF-α的释放,以及白细胞表面Fas配体的激活;两者都可激活Caspase系统,引起细胞凋亡。图中,TNF-α:肿瘤坏死因子-α;Fas-L:Fas配体;CAD:Caspase激活的DNA水解酶;DNA fragmentation:DNA裂解;Apoptosis:凋亡
图1 Caspase家族参与凋亡的过程[8]
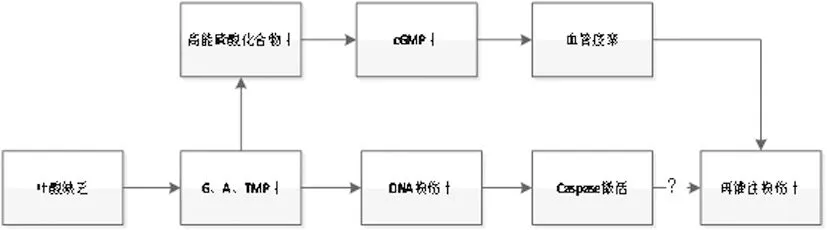
叶酸缺乏引起GMP、AMP和TMP合成障碍,一方面高能磷酸化合物合成减少,cGMP合成减少,影响血管的舒张功能,血管出现痉挛的现象;另一方面,DNA损伤加重而不能修补,Caspase激活引起凋亡,可能加重再灌注损伤。图中cGMP:环磷酸鸟苷;GMP:鸟苷酸;AMP:腺苷酸;TMP:胸苷酸。↑:加重;↓:受抑制;?:表示暂时没有非常明确的文献指出Caspase激活导致再灌注损伤加重
图2 叶酸缺乏导致灌注损伤加重的机制[5-8]
1.3核因子kappa B激活 在心血管和肾脏等疾病中,核因子kappa B( nuclear factor kappa B, NF-κB)上调炎性因子的表达,起到重要的作用[9-10]。NF-κB是一个二聚体蛋白,一方面可以与DNA结合,另一方面还可以与核因子κB抑制蛋白(NF-κB inhibitor, IκB)结合,IκB是NF-κB的抑制蛋白,一般情况下与NF-κB结合成为二聚体,抑制NF-κB的活性。高同型半胱胺酸血症的患者动脉粥样硬化的病理过程中,NF-κB所起到的作用已经基本被阐释清楚了[9]。首先是同型半胱胺酸通过多种途径致使IκB磷酸化并改变结构,与NF-κB脱离。NF-κB游离后进入细胞核内,与某些基因的启动子结合,并调节和促进这些基因表达[11],这些基因中就有MCP-1基因。有研究表明,高同型半胱胺酸血症的大鼠肾脏,其IκB的含量明显低于正常血同型半胱胺酸浓度的大鼠,凝胶电泳的结果发现,NF-κB与DNA结合的比率大大提高[12-13]。用NF-κB的抑制剂预处理高同型半胱胺酸血症的大鼠可以发现,MCP-1的表达明显减少[9-10]。这些结果共同表明,同型半胱胺酸可以激活NF-κB,并促进某些蛋白如MCP-1的表达。
到目前为止一氧化氮合成酶共有3种形式:神经一氧化氮合成酶,可诱导一氧化氮合成酶(inducible nitric oxide synthase, iNOS)和eNOS。iNOS可过量产生NO,NO经过氧化还原反应产生自由基,攻击组织中的蛋白,形成硝基-蛋白质复合物,从而导致组织的损伤。有研究发现,高甲硫氨酸饮食喂养大鼠4周后,大鼠组织中iNOS的mRNA和蛋白表达都明显增加[9]。免疫组织化学的结果显示,高甲硫氨酸饮食的大鼠,其肾脏组织硝基-蛋白质复合物的浓度明显高于正常饮食的大鼠[9]。使用NF-κB抑制剂二硫代氨基甲酸吡咯烷或-乙酰-半胱氨酸[14]后,不仅NF-κB的激活明显减少,而且iNOS mRNA的表达也明显降低(图3)。

叶酸缺乏引起同型半胱氨酸浓度的升高,激活NF-κB,一方面激活MCP-1表达,增加单核细胞的驱化,增加再灌注的损伤;另一方面激活iNOS的表达,过量的NO产生自由基,增加再灌注的损伤。图中,ONOO-和O2-为自由基,↑:上调
图3 NF-κB激活参与叶酸缺乏引起的再灌注损伤[9-11,15]
1.4eNOS解联合与烟酰胺腺嘌呤二核苷酸磷酸的氧化 eNOS、iNOS和nNOS都是二聚体蛋白,它们单体的羧基端都与另一单体的氨基端相衔接,构成氧化还原酶的结构域,这一结构域又与四氢生物蝶呤(tetrahydrobiopteri , BH4)相联合[16]。三种NOS的氧化还原过程如下:第一步,NOS将L-精氨酸氧化成N-羟基-L-精氨酸;第二步,NOS将N-羟基-L-精氨酸氧化成L-瓜氨酸并生成NO。在合成NO的过程中,电子从烟酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate, NADPH)中转移到NO和L-瓜氨酸。如果从NADPH到NO的电子传递链被打断,将造成大量过氧化物的产生,这一过程叫做NOS的解联合[17]。
NADPH依赖的氧化酶是细胞内超氧阴离子自由基的产生主要来源,它由膜结合的组件(GP91PHOX和p22phox亚基)和细胞质组分(p47phox,p67phox,p40phox和Rac1/2亚基)[11,18-20]构成。有研究结果显示,在叶酸缺乏的大鼠模型中,喂养甲硫氨酸12周后,大鼠出现高同型半胱氨酸血症,同时发现NADPH氧化酶的活性明显增高并检测到过氧化阴离子和脂质大量生产。在予以NADPH氧化酶的抑制剂香荚兰乙酮(apocynin)后,检测到NADPH氧化酶的活性降低到正常水平,同时脂质的氧化也被抑制[15]。另外,用同型半胱氨酸培养细胞,对NADPH氧化酶亚基的表达水平进行测量,发现NADHP氧化蛋白4和p22phox亚基的表达显着升高[15,21]。这些证据表明,NADPH在同型半胱氨酸引起的氧化应激中起到了重要的作用。
2 叶酸治疗再灌注损伤
2.1增加高能磷酸化合物 叶酸在调解线粒体功能中起到重要作用。有研究表明,用叶酸喂养实验动物一周,可以提高组织细胞中高能磷酸化合物(包括ADP和ATP)的含量[22]。在有氧环境中,ATP大量合成并保持在一定的浓度水平。在再灌注过程中,ATP由无氧糖酵解产生,因此磷酸肌酸等高能磷酸化合物不能满足组织需要,ATP的净含量也因此下降。用叶酸治疗干预,由于组织储存更多的高能磷酸化合物,可以延缓细胞致死性损伤的发生。用叶酸预处理动物模型已经证明,叶酸可以缩减心肌梗死后的再灌注损伤面积。
2.2增强eNOS与BH4的联合 有研究结果显示,叶酸的活性形式5-甲基四氢叶酸(5-Methyltetrahydrofolate, 5-MTHF)可以增强BH4与eNOS的联合,在5-MTHF的作用下,NO依赖的内皮功能迅速提升,过氧化物的生成明显减少[7]。5-MTHF抑制BH4氧化成BH2,提高BH4的利用,而BH4又可以抑制eNOS的解联合,从而减少过氧化物的产生,起到保护内皮的作用。在这项研究中,实验组予以5-MTHF,对照组予以安慰剂;实验组活性BH4与总蝶呤的比值(BH4/tBio)明显高于对照组。一项相反结果的研究表明,高剂量的叶酸对预防eNOS解联合并未起到作用[23]:首先,高剂量叶酸治疗对高同型半胱氨酸血症的患者有明显效果,但对正常同型半胱氨酸血浓度的患者并不有效[24];其次,虽然叶酸能够减少BH4的氧化并且提高BH4/tBio,但叶酸并不能促进BH4完全氧化后的产物恢复到还原状态[12]。
有研究探究了叶酸与内皮过氧化的关系,有活性的eNOS可以使荧光染料二氢乙啶(Dihydroethidium, DHE)显色,而解联合的eNOS不能使DHE显色。实验组予以5-MTHF,对照组予以安慰剂。用L-NAME(NG-Nitro-L-arginine Methyl Ester),eNOS的抑制剂处理对照组的血管发现,DHE显色明显减弱,说明缺乏叶酸导致eNOS解联合。在实验组中,DHE染色明显强于对照组,说明叶酸可以预防eNOS的解联合[13]。以上这些证据表明叶酸能够保护eNOS,在再灌注中增加内皮功能。
2.3保护内皮 内皮的功能以及白细胞的黏附作用直接与血同型半胱胺酸浓度相关。叶酸不仅降低血中同型半胱胺酸的浓度,从而改善内皮功能,而且在保护内皮的过程中起到独立的作用。有研究表明,将叶酸给与大鼠,不仅血同型半胱胺酸浓度降低,而且MCP-1表达受抑制,单核细胞的黏附作用也受阻[3]。叶酸促进血管内皮eNOS的活性,NO是由eNOS合成的,NO通过激活cGMP促使血管平滑肌舒张;同时,NO能抑制白细胞与血管壁的黏附,NO一方面干扰白细胞黏附因子CD11/CD18与血管内皮的黏附,另一方面抑制白细胞表达CD11/CD18。
另外一个研究用果糖饲喂大鼠,使得大鼠体内BH4含量极低,使其内皮功能受损。对照组仅饲喂果糖,实验组饲喂果糖以及5-MTHF。与对照组相比,实验组大鼠产生更多NO,内皮功能更好,其血管半径相对更大,在再灌注中损伤更小[25]。
最近的一项研究表明,高剂量叶酸治疗和低剂量治疗的疗效观察并没有差别。低剂量的叶酸,相当于食用富含叶酸,可以增加内皮功能并引起内皮功能最大限度地提高。在此基础上再加大叶酸的剂量对血管内皮功能的改善微乎其微[26]。
2.4大规模临床试验表明缺乏证据 虽然在动物模型和体外实验中,叶酸能减少心肌梗死面积,减轻组织的损伤,但一系列临床试验不能得出叶酸能够在梗死中改善缺血的结论。一项北美的大规模临床实验表明,叶酸治疗的结果不尽人意。对无致残性脑梗死的患者观察两年发现,大剂量给与叶酸和小剂量给与叶酸的再次发病率分别为9.2%(166/1814)(RR 1.0, 95%CI 0.8~1.3)和8.8%(161/1835)(RR 1.0,95% CI 0.8~1.3)。在两年的随访中,非致残脑梗死的患者服用叶酸可以降低血同型半胱胺酸浓度,但不能减少脑血管事件再次发生的概率[24]。另一个临床试验Norwegian Vitamin,参与的志愿者共3749例,都是7 d内出现心肌梗死的患者,实验组予以叶酸和维生素B12,对照组予以安慰剂。实验组的同型半胱胺酸浓度低于对照组的27%,但心血管事件的结局并无明显改善。另一实验组予以叶酸、维生素B6和B12,相比对照组,得出一个有害的结论[27]。可能的解释是,B族维生素治疗心肌梗死的效果并不明显,但不良反应已经体现出来了。另外一个临床试验表明,与对照组相比叶酸治疗对心血管疾病和心肌梗死的病死率并不降低[28]。这些临床试验得出一个结论:叶酸治疗缺血性疾病的治疗前景悲观。
虽然临床试验的结果比较悲观,但是实验与临床试验之间有一些不同之处,这些差异造成了两者在结果上的区别。首先,动物实验中,实验组与对照组同型半胱氨酸浓度差异较大,但是临床试验的组间差别并不是很大。其次,在临床试验中,唯一评价的指标是相对危险度。在动物实验中,缺血/再灌注损伤是由动脉栓塞引起的,梗死面积、组织染色被用于作为实验评价的指标。这些指标并不适合在临床试验中使用。
3 结 语
叶酸作为一种重要的维生素,参与了人体的许多重要的生命活动,也在再缺血/再灌注损伤中起到了重要的作用。虽然叶酸缓解再灌注损伤的具体机制不能很清晰地解释清楚,并且叶酸缺乏导致缺血/再灌注损伤的机制也不明,但是叶酸作为一种廉价和实用的治疗药物将会受到热门地研究以及广泛地应用和推广。相信随着科学技术的发展以及研究的进一步深入,学者们终将在叶酸缓解再灌注损伤的具体机制的研究中有所突破。
[1] Bailey SW,Ayling JE.The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake[J].Proc Natl Acad Sci U S A,2009,106(36):15424-15429.
[2] von Eckardstein A,Assmann G.Plasma homocysteine levels and mortality in patients with coronary artery disease[J].N Engl J Med,1997,337(22):1632-1633.
[3] Wang G,Woo CW,Sung FL,et al.Increased monocyte adhesion to aortic endothelium in rats with hyperhomocysteinemia:role of chemokine and adhesion molecules[J].Arterioscler Thromb Vasc Biol,2002,22(11):1777-1783.
[4] Hofmann MA,Lalla E,Lu Y,et al.Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model[J].J Clin Invest,2001,107(6):675-683.
[5] Endres M,Ahmadi M,Kruman I,et al.Folate Deficiency Increases Postischemic Brain Injury[J].Stroke,2005,36(2):321-325.
[6] Endres M,Biniszkiewicz D,Sobol RW,et al.Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase[J].J Clin Invest,113(12):1711-1721.
[7] Antoniades C,Shirodaria C,Warrick N,et al.5-Methyl-tetrahydrofolate rapidly improves endothelial function anddecreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and eNOS coupling[J].Circulation,2006,114(11):1193-1201.
[8] den Hengst WA,Gielis JF,Lin JY,et al.Lung ischemia-reperfusion injury: a molecular and clinical view on a complex pathophysiological process[J].Am J Physiol Heart Circ Physiol,2010,299(5):H1283-1299.
[9] Hwang SY,Woo CW,Au-Yeung KK,et al.Homocysteine stimulates monocyte chemoattractant protein-1 expression in the kidney via nuclear factor-κB activation[J].Am J Physiol Renal Physiol,2008,294(1):F236-244.
[10] Zhang F,Siow YL,O K.Hyperhomocysteinemia activates NF-κB and inducible nitric oxide synthase in the kidney[J].Kidney Int,2004,65(4):1327-1338.
[11] Shiose A,Kuroda J,Tsuruya K,et al.A novel superoxide-producing NAD(P)H oxidase in kidney[J].J Biol Chem,2001,276(2):1417-1423.
[12] Au-Yeung KK,Woo CW,Sung FL,et al.Hyperhomocysteinemia Activates Nuclear Factor-κB in Endothelial Cells via Oxidative Stress[J].Circ Res,2004,94(1):28-36.
[13] Shirodaria C,Antoniades C,Lee J, et al.Global Improvement of Vascular Function and Redox State With Low-Dose Folic Acid: Implications for Folate Therapy in Patients With Coronary Artery Disease[J].Circulation,2007,115(17):2262-2270.
[14] Sung FL,Zhu TY,Au-Yeung KK,et al.Enhanced MCP-1 expression during ischemia/reperfusion injury is mediated by oxidative stress and NF-κB[J].Kidney Int,2002,62(4):1160-1170.
[15] Hwang SY,Siow YL,Au-Yeung KK,et al.Folic acid supplementation inhibits NADPH oxidase-mediated superoxide anion production in the kidney[J].Am J Physiol Renal Physiol,2011,300(1):F189-198.
[16] Frstermann U,Münzel T.Endothelial nitric oxide synthase in vascular disease: from marvel to menace[J].Circulation,2006,113(13):1708-1714.
[17] Warnholtz A,Nickenig G,Schulz E,et al.Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis:evidence for involvement of the renin-angiotensin system[J].Circulation,1999,99(15):2027-2033.
[18] Bokoch GM,Knaus UG.NADPH oxidases: not just for leukocytes anymore![J].Trends Biochem Sci,2003,28(9):502-508.
[19] Gill PS,Wilcox CS.NADPH oxidases in the kidney[J].Antioxid Redox Signal,2006,8(9/10):1597-607.
[20] Griendling KK,Sorescu D,Ushio-Fukai M,et al.NAD(P)H oxidase:role in cardiovascular biology and disease[J].Circ Res,2000,86(5):494-501.
[21] Yang ZZ,Zou AP.Homocysteine enhances TIMP-1 expression and cell proliferation associated with NADH oxidase in rat mesangial cells[J].Kidney Int,2003,63(3):1012-1020.PMID: 12631082
[22] Hwang SY,Siow YL,Au-Yeung KK,et al.High dose folic acid pre-treatment blunts cardiac dysfunction during ischemia coupled to maintenance of high energy phosphates and reduces post-reperfusion injury[J].Circulation,2008,117(14):1810-1819.
[23] Piao CS,Kim DS,Ha KC,et al.The Protective Effect of Epigallocatechin-3 Gallate on Ischemia/Reperfusion Injury in Isolated Rat Hearts:An ex vivo Approach[J].Korean J Physiol Pharmacol,2011,15(5):259-266.
[24] Toole JF,Malinow MR,Chambless LE,et al.Lowering Homocysteine in Patients with Ischemic Stroke to Prevent Recurrent Stroke,Myocardial Infarction,and Death[J].JAMA,2004,291(5):565-575.
[25] Dragoni S,Gori T,Di Stolfo G,et al.Folic Acid Does Not Limit Endothelial Dysfunction Induced by Ischemia and Reperfusion[J].J Cardiovasc Pharmacol,2005,46(4):494-497.
[26] Hyndman ME,Verma S,Rosenfeld RJ,et al.Interaction of 5-methyltetrahydrofolate and tetrahydrobiopterin on endothelial function[J].Am J Physiol Heart Circ Physiol,2002,282(6):H2167-2172.
[27] Williams C,Kingwell BA,Burke K,et al.Folic acid supplementation for 3 wk reduces pulse pressure and large artery stiffness independent of MTHFR genotype[J].Am J Clin Nutr,2005,82(1):26-31.. 16002796
[28] Lonn E,Yusuf S,Arnold MJ,et al.Homocysteine Lowering with Folic Acid and B Vitamins in Vascular Disease[J].N Engl J Med,2006,354(15):1567-1577.16531613
猜你喜欢
基层中医药(2020年5期)2020-09-11 06:32:04
中国生殖健康(2020年6期)2020-02-01 06:28:54
中国化肥信息(2019年12期)2020-01-16 08:40:06
中国生殖健康(2019年12期)2019-01-07 01:54:38
中国生殖健康(2018年6期)2018-11-06 07:09:30
中国化肥信息(2018年7期)2018-08-23 09:12:32
中国化肥信息(2018年6期)2018-08-23 09:11:42
中国化肥信息(2017年7期)2017-12-13 08:46:28
妈妈宝宝(2017年4期)2017-02-25 07:01:16
中国合理用药探索(2012年2期)2012-03-20 16:30:30
